
Abstract
Even tissues capable of complete regeneration, such as bone, show an age-related reduction in their healing capacity. Here, we hypothesized that this decline is primarily due to cell non-autonomous (extrinsic) aging mediated by the systemic environment. We demonstrate that culture of mesenchymal stromal cells (MSCs) in serum from aged Sprague–Dawley rats negatively affects their survival and differentiation ability. Proteome analysis and further cellular investigations strongly suggest that serum from aged animals not only changes expression of proteins related to mitochondria, unfolded protein binding or involved in stress responses, it also significantly enhances intracellular reactive oxygen species production and leads to the accumulation of oxidatively damaged proteins. Conversely, reduction of oxidative stress levels in vitro markedly improved MSC function. These results were validated in an in vivo model of compromised bone healing, which demonstrated significant increase regeneration in aged animals following oral antioxidant administration. These observations indicate the high impact of extrinsic aging on cellular functions and the process of endogenous (bone) regeneration. Thus, addressing the cell environment by, for example, systemic antioxidant treatment is a promising approach to enhance tissue regeneration and to regain cellular function especially in elderly patients.
Similar content being viewed by others


Main
Mesenchymal stromal (or stem) cells (MSCs) and their progeny contribute to the regeneration of mesenchymal tissues, and enable, among others the scarless repair of injured bones.1, 2 This specific regeneration potential arises not only from the MSC ability to differentiate into various mesenchymal cell types to replace cells in damaged tissues but also from paracrine effects of the cells modulating injury or immune responses.3, 4, 5, 6 However, healing capacity of mesenchymal tissues, especially of bone and muscle, has been shown to decline with increasing age.7, 8, 9 In particular, our previous in vivo study showed an age-related delay in the course of bone healing, resulting in an altered microstructure and in reduced mechanical properties of the regenerated tissue.10, 11
Based on the high relevance of MSCs for the mesenchymal tissue regeneration, it is reasonable to presume that this aging phenomenon is at least partially correlated to a decline in the regenerative potential of these cells. Although along with others, we observed no age-dependent change in the differentiation potential of MSCs, our recent functional and proteomic analysis of MSCs derived from young (3 weeks, yMSCs), middle-aged (3 months, mMSCs) and aged (12 months, aMSCs) animals proved an intrinsic (cell autonomous) aging.12, 13 This was associated with a decline in MSC number, reduction of their migration potential and enhanced susceptibility toward senescence.12, 14 Molecular data strongly suggest that these effects of MSC aging are related to an altered cytoskeleton turnover and impaired antioxidant defense.
However, aging is a multifaceted process not only regulated on molecular and cellular, but also on systemic level.15, 16, 17 A minor number of studies address the question of the age-related influence of the systemic environment on cellular function. Conboy et al.18, 19 performed parabiotic pairing experiments between young and aged mice. Results of these experiments suggest that satellite cells and liver progenitor cells of aged mice can be rejuvenated in vivo by exposure to a young systemic milieu. Recently, it was also shown that such heterochronic parabiosis reverses age-related cardiac hypertrophy.20 Thus, we hypothesize that extrinsic (cell non-autonomous) aging has a higher impact on the function of MSCs than intrinsic aging.
To explore potential mechanisms and consequences by which an age-altered systemic environment affects young and aged MSC functions, we studied concurrently cellular and molecular changes in response to serum derived from young and aged Sprague–Dawley rats. Our results show that the systemic environment modulates age-dependent MSC survival and differentiation. Our protein expression and cell assay data identified increased intracellular (oxidative) stress as a potential cause for the altered MSC function. Conversely, antioxidant treatment markedly improved age-altered MSC function and in vivo bone regeneration. In summary, we propose that the systemic environment crucially contributes to the age-related decline in bone regeneration by increasing intracellular ROS levels, hence compromising viability and function of mesenchymal (progenitor) cells.
Results
Age-altered systemic environment reduces proliferation, increases cell cycle inhibitor expression and apoptosis of MSCs
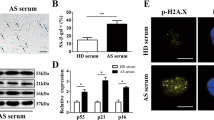
Since our previous results indicate a gradual decline in MSC number and function with aging,12 we used MSCs and serum from 3 weeks (yMSCs; ySerum) to 12 months (aMSCs, aSerum) old male Sprague–Dawley rats for in vitro investigations. To determine the influence of ySerum and aSerum on the growth dynamics of yMSCs and aMSCs, we assessed the number of population doublings (PD) in short-term proliferation assays (Figure 1a). Both yMSCs and aMSCs grown in aSerum displayed significantly reduced proliferation rates compared with the corresponding cultures in ySerum (yMSCs: PDaSera=1.68, PDySera=2.16, P=0.005; aMSCs: PDaSera=1.36, PDySera=1.93, P=0.016). To examine the underlying molecular causes for the reduced cell proliferation in aSerum, the expression of cell cycle inhibitors in yMSCs and aMSCs was analyzed by western blot technique (Figures 1b–d). This analysis revealed a significantly enhanced cyclin-dependent kinase inhibitor 1 (p21WAF1/CIP1) and cyclin-dependent kinase inhibitor 2A (p16INK4A) expression in yMSCs (3.1-fold and 2.6-fold, respectively) and aMSCs (3.0-fold (P=0.004) and 3.1-fold (P=0.004), respectively) cultured in aSerum compared with their counterparts grown in ySerum.

Effects of serum from young (ySerum) and aged (aSerum) Sprague–Dawley rats on proliferation and survival of MSCs. (a) Proliferation rates were determined by using CyQuant assay and obtained values were used to calculate the number of PD. Growth of MSCs from young (yMSCs) and aged (aMSCs) animals cultured in aSerum was significantly lower than in parallel cultures with ySerum. (b and c) Compared with ySerum, MSC culture in aSerum caused an upregulation of the cell cycle inhibitors p21WAF1/CIP1 and p16INK4A. Graphs illustrate quantified signal intensities relative to GAPDH and (d) shows the corresponding representative pictures of the western blot analysis. (e) Apoptosis rates of MSCs cultured in ySerum and aSerum, respectively, were determined by caspase-Glo 3/7 Assay. Graph shows the obtained fluorescence value normalized to total DNA content. (For (a–d), n=6, *P<0.05, Mann–Whitney U-test; (e), n=6, *P<0.05, #P<0.05—positive control (Etoposide) versus all other treatment groups; ANOVA with Bonferroni correction)
To test whether the increased cell cycle inhibitor expression correlates with higher apoptosis rates, we determined the caspase-3/7 activity as surrogate marker for apoptotic cell death,21 and used 50 μM etoposide as a positive control (Figure 1e). Caspase-3/7 activity in yMSCs and aMSCs cultured in aSerum were 39% (P=0.010) and 35% (P=0.006) higher, respectively, than in the parallel cultures with ySerum. Notably, no significant difference in proliferation rate, cell cycle inhibitor expression and apoptosis rate were observed between aMSCs and yMSCs cultured in the same serum.
Age-altered systemic environment affects MSCs osteogenic and adipogenic differentiation capacity
To further investigate the impact of an age-altered systemic environment on the progenitor phenotype of yMSC and aMSCs, we compared their osteogenic and adipogenic differentiation capacity. Osteogenic differentiation was quantified by matrix mineralization using Alizarin Red (AR) and normalized to the number of viable cells (Figures 2a and b). MSCs cultivated in aSerum showed a significantly lower capability to differentiate into the osteogenic lineage (yMSCs: 3.0±0.3; aMSCs: 2.0±0.1) than the parallel cultures with ySerum (yMSCs: 6.0±0.4, P<0.001; aMSCs: 3.6±0.2, P<0.001). In addition, we also found significant differences in matrix mineralization between aMSCs and yMSCs grown in the same serum (ySerum: P<0.001; aSerum: P=0.006).

Serum from aged animals decreases osteogenic potential of MSCs and enhances their differentiation into adipocytes. (a) Osteogenic differentiation was determined by matrix mineralization (AR staining). Shown are representative pictures of yMSCs and aMSCs cultures with OM supplemented with ySerum or aSerum, respectively. (b) Diagrams show optical density values of AR normalized to cell number (osteogenic differentiation index). (c) Representative pictures of MSCs cultured in adipogenic medium supplemented with ySerum or aSerum, respectively. (d) Adipogenic differentiation was quantified by using OR staining and normalized to cell number. (For (a–d), n=6, *P<0.05, #P<0.05—negative control (neg.CTR=expansion medium) versus all other indicated groups; ANOVA with Bonferroni correction)
To examine the effect of an age-altered systemic environment on adipogenic differentiation, we used Oil Red O (OR) staining to quantify lipid vacuoles and normalized obtained values to the number of viable cells (Figures 2c and d). A trend of increased adipogenic differentiation was observed for yMSCs cultured in aSerum (1.34±0.20) compared with their counterparts in ySera (1.11±0.07, P=0.112), but did not reach statistical significance. However, aMSCs cultured in aSerum (1.71±0.14) displayed a significantly higher adipocytes/viable cell ratio compared with their corresponding cultures in ySerum (1.14±0.08, P<0.001) as well as to all other treatment groups. Together these results indicate that age-related changes in the systemic environment reduce MSC proliferation and survival, compromise their osteogenic differentiation ability, and promote their differentiation into the adipogenic lineage.
Age-altered systemic environment alters expression of proteins related to mitochondria, unfolded protein binding and stress response
In order to identify underlying molecular changes responsible for the observed functional alteration in an age-altered systemic environment, we profiled the protein expression of yMSCs and aMSCs cultured in aSerum and ySerum, respectively. Two-dimensional gel electrophoresis detected a total of 147 protein spots to be modulated by the age-altered systemic environment in MSCs. Using mass spectrometry (MS), 105 of these spots were identified as 89 different proteins and 16 isoforms (Supplementary Table 1). Among the detected proteins, 49 were significantly and reproducibly upregulated and 34 downregulated in yMSCs grown in aSerum compared with corresponding cultures in ySerum (Figure 3a). In aMSCs grown in aSerum, 53 proteins were increased and 33 proteins decreased in their expression compared with the parallel cultures with ySerum. Taken together, the expression of 80 proteins were serum-dependently modulated in both MSC groups (nupregulated=48, ndownregulated=32).

Serum-dependent proteomic changes in yMSCs and aMSCs. Two-dimensional gel electrophoresis and MS were used to analyze the protein expression pattern of MSCs cultured in ySerum and aSerum, respectively. (a) Graph depicts the number differentially expressed proteins of MSCs cultured in aSerum compared with corresponding cultures in ySerum. (b) Pie charts show the results of functional annotation clustering according to (left) cellular component and (right) biological process of the serum-dependently expressed proteins. Major clusters are indicated by bold font. (At least three independent experiments with two technical replicates were carried out for all treatment groups)
Subsequently, we performed functional annotation clustering of the differentially expressed proteins according to cellular component, biological process and pathway (Figure 3b). Assessment of cellular component by gene ontology analysis indicated that a large number of proteins were part of an organelle (GO:0044422, ‘organelle part’, n=40, P<0.001), including mitochondria, endoplasmic reticulum and the cytoskeleton (Supplementary Table 2). Clustering according to biological processes revealed that differentially regulated proteins are also related to a ‘cellular metabolic process’ (GO:0044237, n=50, P<0.001) or associated with the ‘response to stress’ (GO:0006950, n=19, P=0.017; Supplementary Table 3).
In particular, proteins within the cluster ‘cellular metabolic process’ are associated with the mitochondria (GO:0005739, n=19, P=0.003), are involved in the ‘generation of precursor metabolites and energy’ (GO:0006091, n=11, P<0.001) or serves as an electron acceptor–donor in the electron transport system (GO:0009055, n=5, P=0.014). Analysis in respect to the pathways revealed that seven of these proteins are part of the glycolysis/gluconeogenesis pathway (P<0.001).
In accordance to the altered expression of proteins of the endoplasmic reticulum (GO:0005783, n=8, P=0.009), the cluster ‘cellular metabolic process’ also contains proteins involved in unfolded protein binding (GO:0051082, n=11, P<0.001). The majority of these proteins are molecular chaperones (n=10, P<0.001). Interestingly, chaperones like the heat shock protein (HSP) 90 kDa beta member 1 (HSP90b1), HSP70 and HSP40, which are known to be upregulated in response to stress, are more highly expressed in MSCs cultured in aSerum. In line with these result, we found that the majority of proteins clustered into the biological process ‘response to stress’ (13 out of 18) were upregulated in response to aSerum. These results led us to assume that the age-altered systemic environment may directly or indirectly provoke enhanced intracellular stress, which would be detrimental to MSC function and survival.
Age-altered systemic environment enhances intracellular oxidative stress and thereby indirectly compromises MSC differentiation
Exemplarily, differentially expressed proteins involved in ‘unfolded protein binding’ were validated by western blot of whole MSC lysates to confirm the proteome data (Figure 4). A significant increase in the expression of HSP90 (yMSCs: 1.3-fold P=0.003; aMSCs: 1.4-fold P=0.002), HSP70 (yMSCs: 1.3-fold P=0.010; aMSCs: 1.6-fold P=0.030), HSP40 (yMSCs: 1.7-fold P=0.012; aMSCs: 1.8-fold P=0.030) and PDI (yMSCs: 1.7-fold P=0.030; aMSCs: 1.7-fold P=0.014) was detected in MSCs cultured in aSerum. In accordance with our proteome analysis, expression of the mitochondrial chaperone HSP60 was significantly decreased in yMSCs (0.6-fold P=0.009) and aMSCs (0.8-fold P=0.010) grown in aSerum.

MSC culture in aSerum modulates the expression of HSP and molecular chaperones. (a) Representative western blots for HSP90, HSP70, HSP60, HSP40, protein disulfide isomerize (PDI), and the corresponding γ-Adaptin control blot. (b) Quantification of western blot analysis depicting the expression level of the indicated protein normalized to γ-Adaptin and relative to corresponding MSCs in ySerum. (For (b), n=6, *P<0.05; Mann–Whitney U-test)
To test whether this enhanced HSP/chaperone expression is correlated with higher levels of intracellular oxidative stress, we incubated yMSCs and aMSCs with the fluorescent ROS indicator CM-H2DCFDA and normalized obtained values to DNA content. MSCs pre-treated with the intracellular ROS generator pyocyanin (500 μM) were used as positive controls (Figures 5a and b). Cultivation of MSCs in aSerum lead to an increased accumulation of ROS compared with the corresponding cells grown in ySerum, indicating enhanced intracellular oxidative stress. Notably, supplementation of the potent antioxidant N-acetyl-L-cysteine (NAC) attenuated the pyocyanin-induced as well as the aSerum-induced ROS generation. Enhanced ROS production has been previously shown to induce global macromolecular damages in cellular constituents like cytosolic proteins, DNA and lipids.22, 23 To elucidate if the aSerum-induced ROS levels are sufficient to cause protein damages, the levels of protein oxidation (carbonyl groups) were determined as a surrogate marker (Figure 5c). In line with the enhanced ROS levels in cells cultured in aSerum, also the levels of carbonyls in yMSCs and aMSCs were 1.7-fold (P=0.013) and 2-fold (P=0.038) higher, respectively. These results strongly suggest that growth of MSCs in aSerum promotes oxidative stress, and further strengthen our assumption that ROS might be the principal cause for the functional alteration in response to an age-altered systemic environment. To validate this hypothesis, we cultivated yMSCs in osteogenic media (OM) supplemented with pyocyanin (5 μM) or a combination of pyocyanin and NAC (2 mM; Figure 5d). Supplementation of pyocyanin significantly reduced matrix mineralization (1.9±0.3) compared with corresponding osteogenic controls without supplements (5.5±0.4, P<0.001). Similar to the results from the ROS measurement, the additional supplementation of NAC could at least partially compensate the pyocyanin effect (3.5±0.4, P<0.007).

Intracellular ROS concentrations and the amount of oxidatively modified proteins are increased in MSC cultures with aSerum. Both, cultures of (a) yMSCs and (b) aMSCs with aSerum displayed higher ROS production than corresponding cultures in ySerum. Supplementation of the antioxidant NAC attenuated the aSerum-induced ROS generation in both MSC populations. Intracellular ROS level were determined using CM-H2DCFDA and normalized to total DNA content. Cell cultures pre-treated with pyocyanin (500 μM) and a combination of pyocyanin and NAC (2 mM) served as controls. (c) Cultivation of MSCs in aSerum results in significantly higher levels of oxidatively modified proteins. ROS-related damages to proteins were analyzed by using OxyBlot Protein Oxidation Detection Kit. Graph shows quantified signal intensities normalized to loading control (γ-Adaptin). (d) To investigate a potential relationship between oxidative stress and altered MSCs differentiation ability, yMSCs were cultured in OM with pyocyanin (5 μM) and/or NAC (10 μM). Graph depicts quantitative analysis of the amount of AR staining normalized to cell number (osteogenic differentiation index) for each treatment group. (For (a–c), n=6, (d) n=3; *P<0.05, #P<0.05—pyocyanin group versus all other treatment groups; (a, b and d) ANOVA with Bonferroni correction; (e) Mann–Whitney U-test)
Oral antioxidant treatment enhances bone regeneration in aged animals
Our in vitro analysis suggested that the age-altered systemic environment compromises MSC function via the induction of intracellular (oxidative) stress, which could be (at least partially) reversed by treatment with the antioxidant NAC. Next, we investigated whether systemic antioxidant administration is capable to improve bone healing in aged Sprague–Dawley rats. In parallel, we monitored NAC therapy in middle-aged animals with suboptimal fixation stiffness, which also delays bone regeneration despite the presence of a biologically competent environment.24, 25
Six weeks after surgery, qualitative radiologic analyses were performed using microcomputed tomography (micro-CT; Figures 6a–f). This analysis revealed that NAC treatment of middle-aged and aged animals results in an increased bone fraction volume (BV/TV) compared with the corresponding untreated controls (P=0.038 and P=0.032, respectively). On the microstructural level, NAC treatment led to a significantly increased trabecular number (Tb.N; P=0.014 and P=0.009, respectively) and reduced trabecular separation (Tb.Sp; P=0.010 and P=0.009, respectively) compared with the untreated controls. No difference in trabecular thickness (Tb.Th) was observed between the regenerated tissue of NAC-treated and untreated animals (P=0.254 and P=0.520, respectively).

Oral NAC administration improved the microstructure and biomechanical quality of the regenerated tissue. NAC therapy was monitored in a rat osteotomy model using aged animals with a rigid fixation stiffness and middle-aged animals with suboptimal (semi-rigid) fixation stiffness. (a) Representative three-dimensional microCT reconstructions from middle-aged Sprague–Dawley with NAC (left, n=8) or without NAC (right, n=6) administration at 6 weeks after surgery. (b) Three-dimensional reconstructions from microCT scans of aged rats with NAC (left, n=8) or without NAC (right, n=7) administration. (c–f) Shown are the results of quantitative microCT evaluation for the indicated age groups with and without NAC treatment. Graphs depict the following morphometric parameters for bone microarchitecture: (c) BV/TV (%), (d) Tb.N (1/mm), (e) Tb.Th (mm) and (f) Tb.Sp (mm). (g–i) Biomechanical testing was performed 6 weeks after surgery. NAC treatment significantly increases (g) MTF, (h) TS and (i) ETF of the regenerated bone tissue of middle-aged and aged animals. (For (c–i), n=6–8; *P<0.05; Mann–Whitney U-test)
To investigate whether the improved mineralization or tissue microstructure of regenerated bone after NAC treatment is also accompanied by an enhanced biomechanical tissue quality, qualitative biomechanical testing was performed (Figures 6g–i). Similar to the microCT data, biomechanical testing showed that NAC treatment significantly increases maximum torque at failure (MTF=36±9 (% contralateral)), torsional stiffness (TS=27±8 (% contralateral)) and energy to failure (ETF=68±19 (% contralateral)) of the regenerated bone of aged animals compared with the untreated group (MTF=12±3% P=0.023; TS=7±3% P=0.025; and ETF=16±6% P=0.013). In addition, also in the middle-aged animal group with suboptimal fixator stiffness, NAC treatment led to superior healing (MTF=60±10%; TS=50±7%; and ETF=54±10) compared with the untreated controls (MTF=25±4% P=0.014; TS=18±4% P=0.010; and EFT=21±4 P=0.014).
Discussion
There is evidence that tissue regeneration is impaired in aged individuals, although details of the underlying mechanisms remain unclear. Moreover, conventional therapies seems to be less effective in aged than in young patients, resulting in a clear demand for new regenerative approaches to overcome the pathophysiological challenges in the elderly.7 Thus, the aspect of aging represents one of the major issues in the field of regenerative medicine. Apart from an incomplete understanding of the exact mechanisms by which certain adult progenitor cells, like MSCs, contribute to regeneration, there is a paucity of data about how growing old influences their regenerative potential.26 In this study, we focused our analyses on bone regeneration as this is one of the few tissues lacking scar formation and allowing complete restoration of initial structure and function under optimal biomechanical and biochemical conditions.27, 28 More importantly, previous investigations showed an age-related decline in bone regeneration, which can be traced back to a decline in both the number and function of bone marrow MSC.12, 29 However, underlying mechanisms for age-associated decrease in MSC regeneration potential are still elusive. In addition to intrinsic alterations, extrinsic systemical factors seem to have an important role in stem cell aging.30
In this study, we showed that MSC proliferation and differentiation in vitro is highly affected by the systemic environment provided by the surrounding serum. MSCs cultured in aSerum exhibited a decreased proliferation and osteogenic differentiation ability and an increased differentiation into adipocytes compared with cells in ySera. These observations were independent from the intrinsic age of the analyzed MSCs. Our results are in concordance with another in vitro study demonstrating a decreased osteoblast differentiation of human MSCs from women in aSera compared with ySera.31
Altered proliferation and differentiation of MSCs could be because of (a) systemic factors in ySera, which directly promote these processes or (b) systemic factors in aSera, which indirectly compromise these processes via increased cellular stress. Enhanced MSC apoptosis and the higher expression of cell cycle inhibitors in response to aSera led us to assume the latter. These observations are in line with the results from other studies showing an excessive in vivo and in vitro expression of cell cycle inhibitors in satellite cells of aged muscle fibers.8 Correspondingly, our proteome analysis revealed altered expression of proteins associated with the mitochondria, protein (re)folding and the general response to stress. Mitochondria are the major source of endogenous ROS and mitochondrial dysfunction leads to elevated oxidative stress levels, which compromise the overall ability of cells to produce and maintain properly folded proteins.32 Thus, enhanced oxidative stress results in accumulation of mis-folded or damaged proteins, which induces the unfolded protein response and enhances the production of molecular chaperones.33, 34, 35 Under normal conditions, molecular chaperones such as HSP90, HSP70, HSP60 and HSP40 have numerous roles in cellular processes, including normal protein folding during translation, refolding of accumulated proteins and the regulation of protein degradation. In response to various environmental stresses, HSPs are upregulated to refold damaged molecules and/or to prevent their irreversible aggregation with other proteins.36, 37 Accordingly, HSPs are abundantly expressed (1–2% of the total protein) in unstressed cells but their levels increase to 4–6% upon proteotoxic stresses.38 In this study, we observed elevated expressions of HSP90, HSP70 and HSP40 in response to an age-altered systemic environment, which coincide with enhanced intracellular ROS levels and increased amounts of oxidatively modified proteins. HSP90 and HSP70 are highly conserved proteins and represent two different classes of molecular chaperone.36 HSP70 prevents irreversible aggregation of unfolded proteins and catalyzes their refolding in an energy- and HSP40-dependent reaction. HSP90 also prevents the aggregation of intermediately folded proteins, but lacks the ability to refold denatured proteins. In addition, HSP90 and HSP70 exhibit anti-apoptotic functions and promote cell survival via inhibiting caspase activation or subsequent cleavage of their substrates.39, 40 However, if HSP response fails to restore correct protein conformation or to degrade these proteins via the proteasomal system, it triggers cell cycle arrest, apoptosis or senescence.34, 41, 42, 43 Interestingly, we found that the expression of HSP60 is reduced in MSCs cultured in aSerum. A similar finding was reported by another study, showing elevated HSP90, HSP60, HSP40 levels but reduced expressions of HSP60 in MSCs upon temperature stress.44 HSP60 primarily localizes to the mitochondrial matrix and is involved in proper folding of newly translated mitochondrial proteins. The role of HSP60 in the process of cell death is controversial, since pro-survival and pro-apoptotic functions have been reported.40, 45 For example, it prevents stress-induced apoptosis of epithelial cells via direct inhibition of caspase 3 and is upregulated in several types of cancer.40, 45 On the other hand, HSP60 promotes proteolytic maturation of caspase 3 and enhances apoptosis in HeLa and Jurkat cells.46, 47 Thus, the function of HSP60 in the process of cell death seems to depend on the apoptotic context and the reduced expression in response to aSera might indicate an altered mitochondrial structure and function. Collectively, our findings of altered HSP expressions, intracellular ROS levels and the amount of oxidatively modified proteins in MSCs provide further evidence for increased intracellular oxidative stress in response to an age-altered systemic environment, which in turn compromises MSC proliferation and differentiation.
The causal relationship between oxidative stress and altered MSCs differentiation ability is further supported by the finding that acute exposure to pyocyanin, known inducer of ROS, significantly reduced matrix mineralization, which could be at least partially reversed by enhanced oxidative stress protection (NAC treatment). Accordingly, other studies also reported that enhanced oxidative stress levels inhibited osteogenic differentiation48, 49 and trigger the adipogenic differentiation of adipose tissue-derived stem cells and skeletal muscle precursors.50, 51 Thus, our in vitro results led us to conclude that age-related changes of the systemic environment have a higher impact on MSCs than intrinsic aging. Irrespective of the cell donor age, aSerum compromises MSC function and survival via enhanced oxidative stress. This mechanism might also affect other mesenchymal cell types required for bone regeneration and may result not only in delayed healing, but also in a limited quality of the regenerated tissue.
Verifying this hypothesis, we subsequently investigated to which extend a systemic antioxidant administration enhance the in vivo bone regeneration in impaired healing situations. We showed that oral treatment with the potent antioxidant NAC is sufficient to improve the mechanical properties and the microstructure of the regenerated bone tissue in aged animals. However, previous studies showed that the long-term bone-healing outcome is not only dependent on the animal age but also significantly influenced by local mechanical conditions. A certain degree of movement within the fracture gap is required for uneventful healing, but high magnitudes of mechanical strain (e.g., as a consequence of a suboptimal fixator stiffness) prolong the initial inflammation phase and delay healing, potentially also mediated via locally enhanced oxidative stress levels.24, 25 Therefore, we used middle-aged rats with semi-ridge fixator system and could show that treatment with NAC is also a sufficient approach to restore appropriate tissue regeneration in a mechanically challenging environment. On the basis of these results, systemic NAC administration seems to be a potent option to prevent poor healing outcomes under both age- and mechanically compromising conditions.
In summary, we propose that the systemic environment crucially contributes to the age-related decline in endogenous (bone) regeneration by increasing intracellular oxidative stress levels, hence compromising viability and function of mesenchymal (progenitor) cells. From the clinical perspective, our study suggests that systemic antioxidant treatment is a promising opportunity to restore the tissue regeneration potential, especially in elderly patients. From the research perspective, our study prompts for the identification of age-related changes in the serum, but the nature of the causative factor(s), for example, whether they are proteins, peptides or small molecules, is still elusive. We currently aim to validate our findings in the human system and to improve our MALDI-MS proteomic methods to elucidate biochemical alteration in serum from young and aged human donors. Further studies have to be performed to validate our findings in the human system and to identify the serum factor(s) causative for the observed cellular and molecular alterations.
Materials and Methods
Ethics statement
All experiments involving the use of animals were in compliance with the policies and principles established by the Animal Welfare Act, the NIH Guide for Care and Use of Laboratory Animals and the National Animal Welfare (TierSchG) and were approved by State Office of Health and Social Affairs Berlin (permit number: T0434/08, T0232/07 and G0091/10).
MSC and serum isolation
Bone marrow and serum were isolated from 3 weeks to 12 months old male Sprague–Dawley rats (Harlan Winkelmann, Eystrup, Germany, www.harlan.com). The serum from 18 animals per age group was combined in one serum pool and each pool was considered as one biological replicate. For all in vitro assays using rat serum, a minimum of six biological replicates in at least three technically independent experiments were performed on separate days.
MSCs were isolated from the bone marrow of the Sprague–Dawley rats and selected by plastic adherence. For cultivation of MSCs, Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies, Darmstadt, Germany) supplemented with 10% fetal calf serum (Biochrom, Berlin, Germany) and 100 U/ml penicillin+100 μg/ml streptomycin was used (expansion medium).14 Culture medium was substituted twice a week and cells were harvested after reaching 70–80% confluence. Cells were subcultured by trypsinization and seeded at 3300 cells/cm2 on uncoated culture dishes. Cell number and viability of trypsinized MSCs were determined using the cell counter CASY TT (Roche, Mannheim, Germany, www.roche-applied-science.com).
Functional assays
All assays were carried out with primary MSCs in passages 2–4. In order to investigate the age-dependent influence of serum on primary MSCs, functional assays were carried out as described below with 10% serum from either young or aged rats.
Proliferation
For short-term proliferation assays, 2000 MSCs/cm2 were seeded onto 96-well plates (96-MTP). Cell number was measured 1 and 4 days after seeding using CyQuant assay (Promega, Mannheim, Germany, www.promega.com) according to the manufacturer’s instructions.
Differentiation
Osteogenic differentiation of confluent MSCs was induced by using OM (DMEM, 10% rat serum, 50 μM ascorbic acid, 10 mM β-glycerophosphate and 0.1 μM dexamethasone).12 The matrix mineralization was visualized with AR staining. Quantification was achieved by measuring the absorbance of AR (optical density AR) that was normalized to number of viable cells determined by AlamarBlue (Life Technologies). If indicated, pyocyanin (5 μM) and NAC (10 μM) were added to the culture medium. Adipogenic differentiation was induced by using adipogenic medium12 (AM; DMEM, 10% rat serum, 2 μM insulin, 100 μM indomethacin, 500 μM isobutylmethylxanthine and 1 μM dexamethasone) and quantified after OR staining, which was normalized to cell number.
Capase-3/7 activity assay
For short-term proliferation assays, 2000 MSCs/cm2 were seeded onto 96-well plates (96-MTP). Capase-3/7 activity was measured after 4 days of culture using Apo-ONE Homogeneous Caspase-3/7 (Promega) according to the manufacturer’s instructions.
Western blot
The Novex system was used according to the Invitrogen NuPAGE protocol. Primary antibodies were mouse(α-rat CDKN2A/p16INK4a), mouse(α-rat CDKN1A/p21WAF1/Cip1) (1 : 1000, Abcam, Cambridge, UK, www.abcam.com) and mouse (α-rat GAPDH) (1 : 7000, Abcam). As secondary antibody goat (α-mouse IgG) peroxidase was utilized. HSPs/chaperones expression was determined using HSP/Chaperone Antibody Sampler Kit according to the supplier’s protocol (New England Biolabs, Frankfurt am Main, Germany, www.neb-online.de). Amount of total oxidized proteins was determined using the OxyBlot Protein Oxidation Detection Kit (Merck Millipore, Darmstadt, Germany, www.merckmillipore.de) according to the manufacturer’s instructions. Bound secondary antibodies were detected using the chemiluminescent substrate ECL (GE Healthcare Piscataway, NJ, USA, http://www.gelifesciences.com). Band intensities were quantified by NIH ImageJ software package (http://rsb.info.nih.gov/ij/).
2D gel electrophoresis and MS
MSCs were cultivated for 5 days in expansion medium containing serum from aged and young rats, respectively. Subsequent to washing cells with phosphate-buffered saline (PBS), they were harvested with PBS (containing 5 mM EDTA and a phosphatase/protease inhibitor cocktail) and by cautious scraping on ice. Cells were washed again with PBS and the remaining buffer was quantitatively removed before shock freezing of cells in liquid nitrogen. Seventy micrograms of protein lysates were separated by 2DE and silver stained as described previously.52 Delta2D (Decodon, Greifswald, Germany, http://www.decodon.com) was used for gel evaluation as earlier described.12, 53 Protein spot of interest were excised from gels and subjected to in-gel tryptic digestion. Tryptic fragments were analyzed by nanoflow high-performance liquid chromatography (nanoHPLC; Proxeon Easy-nLC, Odense, Denmark)/electrospray ionization—MS/MS on a LCQ Deca XP ion trap instrument (Thermo Finnigan, Waltham, MA, USA) and MALDI TOF/TOF on Autoflex III (Bruker Daltonik, Bremen, Germany). Mass spectra were analyzed using our in-house MASCOT software package license version 2.2 (Matrix Science, London, UK) automatically searching the SwissProt database for rate (SwissProt 51.8). MS/MS ion search was performed with this set of parameters: (i) taxonomy: Rattus norvegicus, (ii) proteolytic enzyme: trypsin, (iii) maximum of accepted missed cleavages: 1, (iv) mass value: monoisotopic, (v) peptide mass tolerance 0.8 Da, (vi) fragment mass tolerance: 0.8 Da and (vii) variable modifications: oxidation of methionine and acrylamide adducts (propionamide) on cysteine. Only proteins with individual ions scores >31 or extensive homology (P<0.05), with at least two independent peptides identified were considered. To conduct functional categorizing, all differentially expressed proteins were submitted to the Database for Annotation, Visualization and Integrated Discovery.54 Proteome analysis was done in at least two technically independent experiments with three biologically independent replicates for each MSC/serum combination.
Reactive oxygen species (ROS) measurement
Concentrations of ROS were determined using CM-H2DCFDA (Life Technologies) as described previously.14 Cells were cultivated in expansion media containing either aSera or ySera. Subsequently, cells were incubated with medium supplemented with 10 μM CM-H2DCFDA for 20 min at 37 °C. The resulting fluorescent signal was measured using Infinite 200Pro plate reader (Tecan, Mainz, Germany, www.tecan.com). Cells pre-treated with 500 μM pyocyanin and/or 2 mM NAC for 20 min were used as positive or negative control, respectively. The fluorescent intensities were normalized to the cell number determined by CyQuant.
Animal surgical procedure and NAC treatment
Operations and postoperative care were performed according to a previously published protocol and it used a standardized biomechanically validated external fixation device.55 The experimental model has been previously described11 and is briefly summarized here. Sixteen aged (52 weeks) and 16 middle-aged (12 weeks) Sprague–Dawley rats were divided into subgroups of eight animals depending on the administration of NAC (NAC+/NAC–). Animals were not restricted in weight bearing. Based on the mean drinking volume of 25±3 ml/day (95% CI) of adult Sprague–Dawley rats, as evaluated in about 100 animals of our previous studies,10, 11 a high dosage of NAC56 was administered via freely available drinking water (1.4 g/kg of body weight/day; ACC 600, Hexal AG, Holzkirchen, Germany) in the NAC+ subgroups from surgery until killing. All other animals (NAC–) received plain water for drinking. Daily, well-being and drinking amount were controlled.
Surgical procedure
Using an anterolateral approach, the left femur was osteotomized at the mid-shaft, distracted to a gap of 1.5 mm and externally fixated using a previously described fixation system.55 The distance between fixator and bone (offset) was set to 7.5 mm in the rigid configuration (aged animals) and 15 mm in the semi-rigid (middle-aged rats) configuration. Two-weekly, in general anesthesia, a radiographical examination was performed regarding the occurrence of implant failures and bony consolidation. One aged (untreated group) and two middle-aged (untreated group) animal were excluded from further analysis as a result of infection or implant failure, respectively. Total follow-up of the study was 6 weeks. After animal killing, both femurs were harvested.
3D in vitro micro-CT
Directly after harvesting the osteotomized and contralateral femur, the K-wires were carefully removed and the specimens were placed in cooled (4 °C) PBS solution in acrylic custom-made batch scanning tubes. Subsequently, bones were scanned on a Viva40 micro-CT (Scanco Medical AG, Brüttisellen, Switzerland) and evaluated as described previously,10 except voxel size was selected to be isotropic and fixed at 10.5 μm and the fixed global threshold was 445.5 mg HA/cm3. Using the binarized images, the following morphometric key parameters for bone microarchitecture57 were determined: (i) bone volume fraction (BV/TV (%)—ratio of segmented bone to total volume), (ii) Tb.Th (mm), (iii) Tb.N (1/mm)—average number of trabeculae per unit length, (iv) Tb.Sp (mm)—mean distance between trabeculae. All analyses were performed on the digitally extracted callus tissue using 3D distance techniques (Scanco Software, Scanco Medical AG).58
Biomechanical testing
Immediately after scanning, at least 4 h after killing, both femurs were embedded with methyl methacrylate (Technovit 3040, Heraeus Kulzer, Hanau, Germany) in custom-made casting containers leaving the fracture zone between the inner pin holes and an equivalent zone at the contralateral bones for torsional testing (ElectroForce TestBench LM1, Bose, Framingham, MA, USA) under an axial pre-load of 0.3 N, an adjustment time of 0.1 N/s, and at a torsion speed of 0.5°/s until failure. The MTF, the TS and the ETF of the operated bone was reported as percentage of intact contralaterals.59
Statistical analysis
The SPSS 18.0 software package (SPSS Inc., Chicago, IL, USA) was used for statistical evaluation. If not stated otherwise, results from at least six independent experiments were analyzed for statistical significance using the Mann–Whitney U-test (two groups). Multiple pairwise comparisons were performed by one-way analysis of variance (ANOVA, repeated measures) and P-values were adjusted using Bonferroni’s P-value adjustment multiple comparison procedure. Unless otherwise specified, results are presented as mean±S.E.M. All tests were analyzed two-sided and P<0.05 was regarded as significant.
Abbreviations
- MSCs:
-
mesenchymal stromal cells
- ROS:
-
reactive oxygen species
- PD:
-
population doublings
- p21WAF1/CIP1:
-
cyclin-dependent kinase inhibitor 1
- p16INK4A:
-
cyclin-dependent kinase inhibitor 2A
- OM:
-
osteogenic media
- AM:
-
adipogenic media
- AR:
-
Alizarin Red
- OR:
-
Oil Red O
- MS:
-
mass spectrometry
- HSP:
-
heat shock protein
- micro-CT:
-
microcomputed tomography
- NAC:
-
N-acetyl-L-cysteine
- MTF:
-
maximum torque at failure
- TS:
-
torsional stiffness
- ETF:
-
energy to failure
- BV/TV:
-
bone volume fraction
- Tb.Th:
-
trabecular thickness
- Tb.N:
-
trabecular number
- Tb.Sp:
-
trabecular separation
- ANOVA:
-
analysis of variance
References
Scotti C, Tonnarelli B, Papadimitropoulos A, Scherberich A, Schaeren S, Schauerte A et al. Recapitulation of endochondral bone formation using human adult mesenchymal stem cells as a paradigm for developmental engineering. Proc Natl Acad Sci USA 2010; 107: 7251–7256.
Bianco P, Riminucci M, Gronthos S, Robey PG . Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells 2001; 19: 180–192.
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD et al. Multilineage potential of adult human mesenchymal stem cells. Science 1999; 284: 143–147.
Glaeser JD, Geissler S, Ode A, Schipp CJ, Matziolis G, Taylor WR et al. Modulation of matrix metalloprotease-2 levels by mechanical loading of three-dimensional mesenchymal stem cell constructs: impact on in vitro tube formation. Tissue Eng Part A 2010; 16: 3139–3148.
Park D, Spencer JA, Koh BI, Kobayashi T, Fujisaki J, Clemens TL et al. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell 2012; 10: 259–272.
Reinke S, Geissler S, Taylor WR, Schmidt-Bleek K, Juelke K, Schwachmeyer V et al. Terminally differentiated CD8+ T cells negatively affect bone regeneration in humans. Sci Transl Med 2013; 5: 177ra136.
Gruber R, Koch H, Doll BA, Tegtmeier F, Einhorn TA, Hollinger JO . Fracture healing in the elderly patient. Exp Gerontol 2006; 41: 1080–1093.
Carlson ME, Hsu M, Conboy IM . Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 2008; 454: 528–532.
Zimmermann EA, Schaible E, Bale H, Barth HD, Tang SY, Reichert P et al. Age-related changes in the plasticity and toughness of human cortical bone at multiple length scales. Proc Natl Acad Sci USA 2011; 108: 14416–14421.
Mehta M, Strube P, Peters A, Perka C, Hutmacher D, Fratzl P et al. Influences of age and mechanical stability on volume, microstructure, and mineralization of the fracture callus during bone healing: is osteoclast activity the key to age-related impaired healing? Bone 2010; 47: 219–228.
Strube P, Sentuerk U, Riha T, Kaspar K, Mueller M, Kasper G et al. Influence of age and mechanical stability on bone defect healing: age reverses mechanical effects. Bone 2008; 42: 758–764.
Kasper G, Mao L, Geissler S, Draycheva A, Trippens J, Kühnisch J et al. Insights into mesenchymal stem cell aging: involvement of antioxidant defense and actin cytoskeleton. Stem Cells 2009; 27: 1288–1297.
Stenderup K, Justesen J, Eriksen EF, Rattan SI, Kassem M . Number and proliferative capacity of osteogenic stem cells are maintained during aging and in patients with osteoporosis. J Bone Miner Res 2001; 16: 1120–1129.
Geißler S, Textor M, Kühnisch J, Könnig D, Klein O, Ode A et al. Functional comparison of chronological and in vitro aging: differential role of the cytoskeleton and mitochondria in mesenchymal stromal cells. PLoS One 2012; 7: e52700.
Apfeld J, Kenyon C . Cell nonautonomy of C. elegans daf-2 function in the regulation of diapause and life span. Cell 1998; 95: 199–210.
Blüher M, Kahn BB, Kahn CR . Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 2003; 299: 572–574.
Russell SJ, Kahn CR . Endocrine regulation of ageing. Nat Rev Mol Cell Biol 2007; 8: 681–691.
Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA . Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 2005; 433: 760–764.
Wagers AJ, Conboy IM . Cellular and molecular signatures of muscle regeneration: current concepts and controversies in adult myogenesis. Cell 2005; 122: 659–667.
Loffredo FS, Steinhauser ML, Jay SM, Gannon J, Pancoast JR, Yalamanchi P et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell 2013; 153: 828–839.
Porter AG, Jänicke RU . Emerging roles of caspase-3 in apoptosis. Cell Death Differ 1999; 6: 99–104.
Finkel T, Holbrook NJ . Oxidants, oxidative stress and the biology of ageing. Nature 2000; 408: 239–247.
Beckman KB, Ames BN . The free radical theory of aging matures. Physiol Rev 1998; 78: 547–581.
Schmidt-Bleek K, Schell H, Schulz N, Hoff P, Perka C, Buttgereit F et al. Inflammatory phase of bone healing initiates the regenerative healing cascade. Cell Tissue Res 2012; 347: 567–573.
Schmidt-Bleek K, Schell H, Lienau J, Schulz N, Hoff P, Pfaff M et al. Initial immune reaction and angiogenesis in bone healing. J Tissue Eng Regen Med 2012 e-pub ahead of print 11 April 2012 doi:10.1002/term.1505.
Sharpless NE, DePinho RA . How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol 2007; 8: 703–713.
Klein P, Schell H, Streitparth F, Heller M, Kassi JP, Kandziora F et al. The initial phase of fracture healing is specifically sensitive to mechanical conditions. J Orthop Res 2003; 21: 662–669.
Gerstenfeld LC, Cullinane DM, Barnes GL, Graves DT, Einhorn TA . Fracture healing as a post-natal developmental process: molecular, spatial, and temporal aspects of its regulation. J Cell Biochem 2003; 88: 873–884.
D'Ippolito G, Schiller PC, Ricordi C, Roos BA, Howard GA . Age-related osteogenic potential of mesenchymal stromal stem cells from human vertebral bone marrow. J Bone Miner Res 1999; 14: 1115–1122.
Rando TA . Stem cells, ageing and the quest for immortality. Nature 2006; 441: 1080–1086.
Abdallah BM, Haack-Sorensen M, Fink T, Kassem M . Inhibition of osteoblast differentiation but not adipocyte differentiation of mesenchymal stem cells by sera obtained from aged females. Bone 2006; 39: 181–188.
Rana A, Rera M, Walker DW . Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc Natl Acad Sci USA 2013; 110: 8638–8643.
Muller M . Cellular senescence: molecular mechanisms, in vivo significance, and redox considerations. Antioxid Redox Signal 2009; 11: 59–98.
Sastre J, Pallardó FV, Viña J . Mitochondrial oxidative stress plays a key role in aging and apoptosis. IUBMB Life 2000; 49: 427–435.
Szweda PA, Friguet B, Szweda LI . Proteolysis, free radicals, and aging. Free Radic Biol Med 2002; 33: 29–36.
Nollen EA, Morimoto RI . Chaperoning signaling pathways: molecular chaperones as stress-sensing 'heat shock' proteins. J Cell Sci 2002; 115: 2809–2816.
Dukan S, Farewell A, Ballesteros M, Taddei F, Radman M, Nyström T . Protein oxidation in response to increased transcriptional or translational errors. Proc Natl Acad Sci USA 2000; 97: 5746–5749.
Garrido C, Gurbuxani S, Ravagnan L, Kroemer G . Heat shock proteins: endogenous modulators of apoptotic cell death. Biochem Biophys Res Commun 2001; 286: 433–442.
Takayama S, Reed JC, Homma S . Heat-shock proteins as regulators of apoptosis. Oncogene 2003; 22: 9041–9047.
Chandra D, Choy G, Tang DG . Cytosolic accumulation of HSP60 during apoptosis with or without apparent mitochondrial release: evidence that its pro-apoptotic or pro-survival functions involve differential interactions with caspase-3. J Biol Chem 2007; 282: 31289–31301.
Franco MC, Ye Y, Refakis CA, Feldman JL, Stokes AL, Basso M et al. Nitration of Hsp90 induces cell death. Proc Natl Acad Sci USA 2013; 110: E1102–E1111.
Friguet B . Protein repair and degradation during aging. Scientific World J 2002; 2: 248–254.
Zhao L, Ackerman SL . Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol 2006; 18: 444–452.
Stolzing A, Sethe S, Scutt AM . Stressed stem cells: temperature response in aged mesenchymal stem cells. Stem Cells Dev 2006; 15: 478–487.
Zhang L, Pelech S, Uitto VJ . Bacterial GroEL-like heat shock protein 60 protects epithelial cells from stress-induced death through activation of ERK and inhibition of caspase 3. Exp Cell Res 2004; 292: 231–240.
Samali A, Cai J, Zhivotovsky B, Jones DP, Orrenius S . Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J 1999; 18: 2040–2048.
Xanthoudakis S, Roy S, Rasper D, Hennessey T, Aubin Y, Cassady R et al. Hsp60 accelerates the maturation of pro-caspase-3 by upstream activator proteases during apoptosis. EMBO J 1999; 18: 2049–2056.
Mody N, Parhami F, Sarafian TA, Demer LL . Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic Biol Med 2001; 31: 509–519.
Chen CT, Shih YR, Kuo TK, Lee OK, Wei YH . Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 2008; 26: 960–968.
Aguiari P, Leo S, Zavan B, Vindigni V, Rimessi A, Bianchi K et al. High glucose induces adipogenic differentiation of muscle-derived stem cells. Proc Natl Acad Sci USA 2008; 105: 1226–1231.
De Pauw A, Tejerina S, Raes M, Keijer J, Arnould T . Mitochondrial (dys)function in adipocyte (de)differentiation and systemic metabolic alterations. Am J Pathol 2009; 175: 927–939.
Klose J, Kobalz U . Two-dimensional electrophoresis of proteins: an updated protocol and implications for a functional analysis of the genome. Electrophoresis 1995; 16: 1034–1059.
Mao L, Zabel C, Wacker MA, Nebrich G, Sagi D, Schrade P et al. Estimation of the mtDNA mutation rate in aging mice by proteome analysis and mathematical modeling. Exp Gerontol 2006; 41: 11–24.
Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol 2003; 4: P3.
Strube P, Mehta M, Putzier M, Matziolis G, Perka C, Duda GN . A new device to control mechanical environment in bone defect healing in rats. J Biomech 2008; 41: 2696–2702.
Shankar K, Hidestrand M, Liu X, Chen JR, Haley R, Perrien DS et al. Chronic ethanol consumption inhibits postlactational anabolic bone rebuilding in female rats. J Bone Miner Res 2008; 23: 338–349.
Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R . Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res 2010; 25: 1468–1486.
Hildebrand T, Rüegsegger P . A new method for the model-independent assessment of thickness in three-dimensional images. J Microsc 1997; 185: 67–75.
White AA, Panjabi MM, Southwick WO . The four biomechanical stages of fracture repair. J Bone Joint Surg Am 1977; 59: 188–192.
Acknowledgements
We thank M Princ as well as L Schumann for excellent technical assistance. This work was supported by grants from the BCRT and BSRT through funding by the German Federal Ministry of Education and Research (BMBF), and by young investigator grant (GE2512/1-1) from the German Research Foundation (DFG) to SG. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict interest.
Additional information
Edited by RA Knight
Author contributions
SG, MT, GK, GND and PS designed research; SG, MT, OK, KS-B, AE, DJ and PS performed research; SG, MT, KS-B, DJ, AO, JK, AD, GND and PS analyzed data, SG, MT, AE, DJ and GK contributed reagents/materials/analysis tools and SG wrote paper.
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Geißler, S., Textor, M., Schmidt-Bleek, K. et al. In serum veritas—in serum sanitas? Cell non-autonomous aging compromises differentiation and survival of mesenchymal stromal cells via the oxidative stress pathway. Cell Death Dis 4, e970 (2013). https://doi.org/10.1038/cddis.2013.501
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.501
Keywords
This article is cited by
-
Individual immune cell and cytokine profiles determine platelet-rich plasma composition
Arthritis Research & Therapy (2023)
-
Unraveling local tissue changes within severely injured skeletal muscles in response to MSC-based intervention using MALDI Imaging mass spectrometry
Scientific Reports (2018)
