
Abstract
The homolog of p53 gene, p63, encodes multiple p63 protein isoforms. TAp63 proteins contain an N-terminal transactivation domain similar to that of p53 and function as tumor suppressors; whereas ΔNp63 isoforms, which lack the intact N-terminal transactivation domain, are associated with human tumorigenesis. Accumulating evidence demonstrating the important roles of p63 in development and cancer development, the regulation of p63 proteins, however, is not fully understood. In this study, we show that peptidyl-prolyl isomerase Pin1 directly binds to and stabilizes TAp63α and ΔNp63α via inhibiting the proteasomal degradation mediated by E3 ligase WWP1. We further show that Pin1 specifically interacts with T538P which is adjacent to the P550PxY543 motif, and disrupts p63α–WWP1 interaction. In addition, while Pin1 enhances TAp63α-mediated apoptosis, it promotes ΔNp63α-induced cell proliferation. Furthermore, knockdown of Pin1 in FaDu cells inhibits tumor formation in nude mice, which is rescued by simultaneous knockdown of WWP1 or ectopic expression of ΔNp63α. Moreover, overexpression of Pin1 correlates with increased expression of ΔNp63α in human oral squamous cell carcinoma samples. Together, these results suggest that Pin1-mediated modulation of ΔNp63α may have a causative role in tumorigenesis.
Similar content being viewed by others

Main
p63 is a member of the p53 gene family, which consists of 15 exons, and contains two transcriptional start sites, a 5′ promoter that precedes the first exon encoding the full transactivation domain (TAD) on the N-terminus, and a cryptic 3′ intronic promoter that gives rise to ΔN isoforms lacking a full TAD. Both TA and ΔN isotypes can undergo alternative splicing to generate different carboxy-termini (α, β, γ), thus giving rise to at least six different p63 isoforms (TAα, TAβ, TAγ and ΔNα, ΔNβ, ΔNγ). Each p63 isoform possesses a DNA-binding domain and an oligomerization domain. In addition, p63α contains a full-length C-terminus, which consists of a sterile alpha motif (SAM) for protein–protein interaction and a trans-inhibitory domain (TID), whereas p63β and p63γ isoforms have truncated C-termini due to alternative splicing.1, 2
The TAp63 isoforms are potent transactivators of a subset of genes, which in part overlap with p53 downstream targets including Bax, Puma, and p21. Consequently, TAp63s can induce both cell cycle arrest and apoptosis. By contrast, ΔNp63 isoforms can also transactivate a subset of genes involved in a variety of biological activities. Importantly, ΔNp63α has been shown to repress transcriptional activity of p53 family members, enabling ΔNp63α to promote cell proliferation and tumorigenesis under certain circumstances.2
Pin1 is a ubiquitously expressed peptidyl-prolyl isomerase, consisting of an N-terminal WW domain and a C-terminal PPIase domain. The WW domain functions as the specific binding domain for Pin1 substrates and selectively binds to phospho-Ser-Pro (pSP) or phospho-Thr-Pro (pTP) motifs.3 Point mutations in the WW domain (W34A, Y23A) of Pin1 abolish the protein–protein interaction between Pin1 and its substrates. After binding to its substrates, Pin1 can facilitate the cis-trans isomerization of pSP/pTP peptidyl-prolyl bonds through its PPIase domain, resulting in conformational and functional changes of substrate proteins.4 It is well documented that Pin1 has important roles in diverse cellular processes, and is overexpressed in diverse human tumors and promotes oncogenesis by modulating numerous proteins involved in tumorigenesis.5, 6, 7
Although the association between p63 and Pin1 has been previously reported, the underlying mechanism and physiological effects of this physical interaction remain largely unclear.8 In this study, we demonstrate that Pin1 binds to TAp63α and protects it from proteasomal degradation, leading to increased apoptosis. On the other hand, Pin1 can also bind to ΔNp63α and inhibits its degradation. Knockdown of Pin1 in FaDu cells lead to a decrease in ΔNp63α protein levels and an inhibition of cell proliferation. Our study reveals a mechanism enabling Pin1 to modulate specific p63 isoforms to regulate cell survival/proliferation and tumorigenesis.
Results
Pin1 interacts with p63 in vitro and in vivo
Pin1 has been shown to physically interact with p53 and p73.9, 10, 11 To test if it can bind to p63 proteins, we performed GST–Pin1 pull-down experiment. Our results showed that TAp63α (Figure 1a) TAp63γ (Figure 1b) and ΔNp63 proteins (Figure 1c) were readily pulled down with GST–Pin1, but not with GST alone.

p63 proteins physically interact with Pin1. (a) and (b) Pin1 interacts with TAp63 in vitro. H1299 cells transfected with either TAp63α or TAp63γ expression plasmid were lysed and subjected to GST pull-down assay with GST alone (lane 2) or GST–Pin1 (lane 3). 10 μg of total protein from the same cell lysates were directly loaded as input controls (lane 1). Immunoblotting analysis (IB) was performed using an antibody specific for p63 (4A4, α-p63; top panels). Comparable amounts of GST and GST–Pin1 fusion proteins were shown by staining the membrane with Coomassie blue R-250 (bottom panels). (c) Pin1 interacts with ΔNp63 in vitro. Pull-down experiments were performed as described above. 10 μg of total protein from lysate inputs (top panel) and pull-down products (bottom panel) were subjected to IB analysis with α-p63. (d) The WW domain of Pin1 mediates Pin1–p63 interaction, which can be abolished by W34A or Y23A point mutation in the WW domain of Pin1. GST pull-down experiments were performed with GST alone (lane 2), GST–Pin1 (lane 3), GST–PPIase (lane 4), GST–WW (lane 5), GST–Pin1 (W34A) (lane 6) or GST–Pin1 (Y23A) (lane 7). (e) Wild-type but not W34A mutant Pin1 can form a stable complex with TAp63α. Lysates from H1299 cells transiently expressing HA-tagged wild-type or W34A mutant Pin1 were subjected to immunoprecipitation (IP) and IB using myc or HA antibody. (f) Endogenous proteins of ΔNp63α and Pin1 form a stable complex in FaDu cells. 2 mg of FaDu cell lysate was subjected to IP with α-p63 (4A4) or IgG control. The IP products were subjected to IB with α-p63 or α-Pin1
It was reported that the physical interactions between Pin1 and its substrates is mediated by the WW domain, but not the PPIase domain of Pin1. Point mutations in the WW domain (Y23A or W34A) completely abolish the interaction between Pin1 and its substrates.4 As shown in Figure 1d, TAp63α bound to the full length or the WW domain of Pin1, but not the PPIase domain, Pin1(W34A), or Pin1(Y23A). In addition, our results of GST–Pin1 pull-down experiment in combination with calf intestine phosphatase and phosphatase inhibitor treatment showed that the interaction between p63 and Pin1 was dependent on p63 phosphorylation (Supplementary Figure S1).
Next, we further examined whether p63 proteins could physiologically interact with Pin1. As shown in Figure 1e, TAp63α formed a stable complex with Pin1, but not with Pin1(W34A), in transiently transfected H1299 cells. To examine the in vivo interaction between Pin1 and ΔNp63α, we performed immunoprecipitation using lysate of the head and neck squamous cell carcinoma (HNSCC) cell line FaDu, which expresses high levels of endogenous Pin1 and ΔNp63α. As shown in Figure 1f, endogenous Pin1 and ΔNp63α form a stable complex in FaDu cells.
Taken together, our data demonstrate that p63 proteins, including TAp63α, TAp63γ, ΔNp63α, and ΔNp63γ,can interact with Pin1, whereas W34 and Y23 in the WW domain of Pin1 are critical for its interaction with p63 proteins.
Pin1 specifically stabilizes p63α,but not p63γ
Since Pin1 was reported to stabilize p73 proteins,9 we asked if Pin1 is involved in regulating p63 protein stability. To this end, we transiently co-expressed TAp63α and Pin1 in H1299 cells. The results of immunoblotting showed that expression of wild-type Pin1, but not Pin1(W34A), led to increased TAp63α protein levels; as a control of transfection efficiency, GFP protein levels were not affected (Figure 2a). In addition, we found that transient expression of Pin1, but not Pin1 (W34A), in HEK293 cell line stably overexpressing ΔNp63α (HEK293:ΔNp63α), upregulates the protein level of ΔNp63α (Figure 2b).

Pin1 upregulates protein levels of p63α, but not p63γ, via increasing their half-lives. (a) Transient overexpression of wild-type but not W34A mutant Pin1, upregulates the protein level of TAp63α in H1299 cells. 1 μg HA-tagged wild-type or W34A mutant Pin1 expression plasmid, or vector control, was transfected together with 200 ng myc-tagged TAp63α expression plasmid and 50 ng pEGFP-N1, into H1299 cells. 48 h after transfection, cells were lysed and subjected to IB. (b) Transient overexpression of wild-type but not W34A mutant Pin1, upregulates the protein level of ΔNp63α in HEK293 cells. 1 μg HA-tagged wild-type or W34A mutant Pin1 expression plasmid, or vector control, was transfected into a HEK293 cell line stably overexpressing ΔNp63α HEK293:ΔNp63α. Forty-eight hours after transfection, cells were lysed and subjected to IB analysis. (c) and (d) Stable expression of wild-type Pin1 upregulates protein levels of TAp63α and ΔNp63α, but not TAp63γ or ΔNp63γ. TAα, ΔNα, TAγ and ΔNγ isoforms of p63 expression plasmids (200 ng each) were respectively co-transfected with 50 ng pEGFP-N1 into H1299 cells stably expressing wild-type (H1299:Pin1) or W34A mutant [H1299:Pin1(W34A)] Pin1. Forty-eight hours after transfection, cells were lysed and subjected to IB analysis. (e) SiRNA-mediated knockdown of Pin1 downregulates protein levels of ΔNp63α in HEK293 cells. SiPin1 or scrambled control (20 pmol each) was transiently transfected into HEK293 cells stably overexpressing ΔNp63α. Forty-eight hours after transfection, cells were lysed and subjected to IB analysis. (f) and (g) Pin1 extends protein half-life of TAp63α. H1299 cells were co-transfected with TAp63α (200 ng) and pEGFP-N1 (50 ng), together with HA-Pin1 (400 ng) or its vector control, for 24 h and then treated with 50 mg/ml cycloheximide (CHX) for indicated durations followed by IB analysis (f). Protein levels of TAp63α were normalized with Actin, and the protein half-life was quantified (g). (h) and (i) Pin1 extends protein half-life of ΔNp63α. HEK293:ΔNp63α cells were transfected with HA-Pin1 (1 μg) or its vector control for 24 h and then treated with 50 mg/ml cycloheximide (CHX) for indicated durations followed by IB analysis (h). The half-life of ΔNp63α proteins was quantified (i)
To further investigate Pin1–p63 interaction, we generated H1299 cells stably expressing wild-type Pin1 (H1299:Pin1) or W34A mutant Pin1 [H1299:Pin1(W34A)], which cannot interact with p63. As shown in the bottom panel of Figure 2c, these two stable cell lines express comparable levels of exogenous wild-type or W34A mutant Pin1. After transfection with p63α or p63γ expression plasmids, H1299:Pin1 cells yielded much higher levels of TAp63α or ΔNp63α than H1299:Pin1(W34A) cells did (Figures 2c and d, lane 2 versus lane 1), while TAp63γ or ΔNp63γ protein levels in both cell lines were comparable (Figures 2c and d, lane 4 versus lane 3); as a control, protein levels of GFP were shown in different transfections.
We next knocked down endogenous Pin1 using Pin1-specific siRNA. As shown in Figure 2e, siRNA-mediated knockdown of Pin1 in HEK293:ΔNp63α cells significantly downregulated ΔNp63α; Pin1 knockdown in H1299 cells also significantly reduced TAp63α and ΔNp63α, but not TAp63γ (Supplementary Figure S2). Given that the p63 and GFP expression plasmids used in our experiments were driven by the same CMV promoters, these data suggest that Pin1 likely upregulates TAp63α and ΔNp63α at post-transcriptional levels.
To examine whether Pin1 affects p63 protein stability, we measured the half-life of TAp63α protein in H1299 cells by means of cycloheximide (CHX) treatment. As shown in Figures 2f and g, Pin1 extended the half-life of TAp63α protein from 1.2 h to 2.2 h; by contrast, the stability of GFP was not significantly affected by Pin1 under similar settings. Data from subsequent experiments with stable H1299 cell lines expressing wild-type or W34A mutant Pin1 showed that TAp63α has a longer half-life in H1299:Pin1 cells than in H1299:Pin1(W34A) cells (Supplementary Figure S3). With HEK293:ΔNp63α stable cells, we found that Pin1 extended the half-life of stably expressed ΔNp63α from about 4 h to approximately 7 h (Figures 2h and i). Taken together, these data suggest that Pin1 binds to and stabilizes p63α proteins.
T538 is critical for Pin1-mediated TAp63α protein stabilization
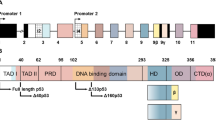
Since Pin1 stabilizes p63α, but not p63γ proteins, we speculated that some regions unique to p63α are required for Pin1 effect on p63α. As depicted in Figure 3a, p63α isoforms possess a unique C-terminal domain, containing six putative Pin1-binding motifs, which are absent in the alternatively spliced p63γ isoforms; among these six Ser/Thr-Pro motifs, T538-Pro is adjacent to the P540PxY543 motif, which was reported as the binding site for ubiquitin E3 ligase WWP1.12 To identify which site is critical for Pin1-mediated stabilization of TAp63α, several single point mutations of TAp63α, including S463A, T491A, T538A, and T619A, were constructed. Wild-type TAp63α or mutant derivatives were transiently co-transfected into H1299:Pin1 or H1299:Pin1(W34A) cells. As shown in Figure 3b, wild-type TAp63α was significantly upregulated by wild-type Pin1; T538A, but not other point mutations, abolished the Pin1-mediated upregulation of TAp63α.

T538A point mutation abrogates Pin1-mediated upregulation and siPin1-mediated downregulation of TAp63α. (a) Schematic representation of putative Pin1-binding sites in different p63 isoforms. TAD, transactivation domain; DBD, DNA-binding domain; OLD, oligomerization domain; SAM, sterile alpha motif; TID, trans-inhibitory domain. Dots indicate putative Pin1-binding sites, which are Serine-Proline (SP) or Threonine-Proline (TP) motifs. A PPxY motif (framed letters) is adjacent to the T538-P in TAp63α or the T444-P in ΔNp63α. (b) T538A, but not other point mutations of putative Pin1-binding sites in the special C-terminus of TAp63α, abrogates the Pin1-mediated upregulation of TAp63α. S463A, T491A, T538A, or T619A mutant, or wild-type TAp63α plasmid (200 ng each) was co-transfected pEGFP-N1 (50 ng) into H1299:Pin1(W34A) or H1299:Pin1 cells. Forty-eight hours after transfection, cells were lysed and subjected to immunoblotting analysis. The relative level of each mutant TAp63α in Pin1(W34A) control was respectively set as 1.0. (c) SiRNA-mediated knockdown of Pin1 downregulates wild-type but not T538A mutant TAp63α Pin1 siRNA (siPin1) or scrambled control (20 pmol each) was transiently co-transfected into H1299 cells with expression plasmid of wild-type (WT) or T538A mutant TAp63α (1 μg each), together with 50 ng pEGFP-N1. Forty-eight hours after transfection, cells were lysed and subjected to immunoblotting analysis. The relative level of wild-type or T538A mutant TAp63α in scrambled control was respectively set as 1.0
To confirm the importance of T538 in Pin1-mediated regulation of TAp63α, we examined the protein levels of TAp63α(T538A) in H1299 cells in which Pin1 was simultaneously knocked down with specific siRNA. As shown in Figure 3c, knockdown of Pin1 led to downregulation of wild-type TAp63α, but not TAp63α (T538A).
These data suggest that T538P, which is adjacent to a P540PxY543 motif, is involved in Pin1-mediated regulation of TAp63α stabilization.
Pin1 inhibits WWP1-dependent degradation of p63α proteins
To investigate whether proteasome-dependent protein degradation is involved in Pin1-mediated upregulation of p63α proteins, we treated cells with a specific proteasome inhibitor MG132 or the vehicle control, DMSO. As shown in Figure 4a, wild-type Pin1, but not Pin1(W34A), increased TAp63α protein levels (lane 2 versus lane 1); protein levels of TAp63α in MG132 treated cells were much higher than in DMSO controls (lane 3 versus lane 1, lane 4 versus lane 2); however, Pin1 failed to further increase TAp63α protein levels in the presence of MG132 (lane 4 versus lane 3). In addition, MG132 was able to reverse the reduction in TAp63α protein levels following Pin1 knockdown (Figure 4b). However, the transfection control GFP, which is not targeted for proteasomal degradation,13 was not affected by MG132 treatment and Pin1 expression. These data suggest that Pin1 inhibits proteasome-mediated degradation of p63α proteins.

Pin1 inhibits proteasome- and WWP1- dependent degradation of p63α proteins. (a) MG132 abrogates Pin1-mediated upregulation of TAp63α H1299:Pin1 or H1299:Pin1(W34A) cells were co-transfected with TAp63α (400 ng) and pEGFP-N1 (50 ng) for 24 h. Cells were then treated with 40 μM MG132, which is a specific proteasome inhibitor, or its vehicle control (0.4% DMSO), for additional 18 h before collection. Cell lysates were subjected to immunoblotting analysis. (b) MG132 abrogates siPin1-mediated downregulation of TAp63α (400 ng). Pin1 siRNA (siPin1) or scrambled control (20 pmol each) was transiently co-transfected into H1299 cells with expression plasmid of TAp63α (1 μg) and pEGFP-N1 (50 ng). Twenty-four hours later, cells were treated with 40 μM MG132 or 0.4% DMSO for an additional 18 h. Then cells were collected and subjected to immunoblotting analysis. (c) WWP1 downregulates TAp63α while wild-type Pin1 can rescue this downregulation. TAp63α expression plasmid (400 ng) was co-transfected with WWP1 (200 ng) or (and) wild-type or W34A mutant HA-Pin1 (1 μg) expression plasmids or their vector controls, together with pEGFP-N1 (50 ng). Forty-eight hours after transfection, cells were collected for immunoblotting analysis. (d) WWP1 downregulates ΔNp63α while Pin1 can rescue this downregulation. WWP1 (200 ng) or (and) HA-Pin1 (1 μg) expression plasmids or their vector controls were co-transfected into HEK293 cells stably overexpressing ΔNp63α (HEK293:ΔNp63α). Forty-eight hours after transfection, cells were collected for immunoblotting analysis. (e) Knockdown of WWP1 upregulates TAp63α and removes the siPin1-mediated downregulation of TAp63α. SiPin1 and (or) siWWP1 (20 pmol each) were co-transfected into H1299 cells together with TAp63α (500 ng) and pEGFP-N1 (50 ng). Forty-eight hours after transfection, cells were collected for immunoblotting analysis. (f) WWP1 downregulates T538A mutant TAp63α but Pin1 cannot rescue this downregulation. T538A mutant TAp63α expression plasmid (400 ng) was co-transfected with WWP1 (200 ng) or (and) HA-Pin1 (1 μg) expression plasmids or its vector control, together with pEGFP-N1 (50 ng). Forty-eight hours after transfection, cells were collected for immunoblotting analysis. (g) Pin1 impairs the binding of WWP1 to wild-type but not T538A mutant TAp63α. 0.5 μg wild-type or T538A mutant Myc-TAp63α plasmid was transfected into H1299:Pin1(W34A) or H1299:Pin1 cells, together with 0.5 μg WWP1 expression plasmid. Twenty-four hours after transfection, cells were lysed and 1 mg total protein samples were respectively subjected to immunoprecipitation with myc antibody (α-myc). The IP products were subjected to immunoblotting analysis
WWP1 has been reported to function as a ubiquitin E3 ligase that promotes proteasome-dependent degradation of p63α proteins.12 Our above-mentioned data indicate that Thr538-Pro, which is adjacent to the WWP1 binding site (PPxY motif), is critical to Pin1-mediated regulation of p63α stability (Figure 3). As shown in Figure 4c, overexpression of WWP1 led to a significant decrease in TAp63α, which was prevented by co-expression of wild-type Pin1, but not Pin1(W34A). Similarly, Pin1 also significantly inhibited WWP1-mediated downregulation of ΔNp63α (Figure 4d). As shown in Figure 4e, knockdown of WWP1 increased TAp63α, whereas knockdown of Pin1 led to decrease in TAp63α; simultaneous knockdown of WWP1 abolished the downregulation of TAp63α mediated by Pin1 knockdown. These data suggest that Pin1 is involved in WWP1-mediated degradation of p63α.
Since we have shown that T538 is critical for Pin1-mediated TAp63α stabilization (Figures 3b and c), we next investigated the effect of WWP1 on TAp63α(T538A) protein stability. As shown in Figure 4f, WWP1 also decreased protein levels of TAp63α(T538A), suggesting that T538A substitution does not affect WWP1-mediated proteasomal degradation; however, overexpression of Pin1 failed to rescue WWP1-mediated downregulation of TAp63α(T538A), though T538A mutation abolished Pin1-mediated stabilization of wild-type TAp63α (Figures 3b and c), suggesting that this substitution removes the binding of Pin1, but not of WWP1, to this region. Further study demonstrated that Pin1 impairs TAp63α–WWP1 but not TAp63α(T538A)–WWP1 interaction: as shown in Figure 4g, though the amount of immunoprecipitated wild-type Myc-TAp63α from H1299:Pin1 was much higher than that from H1299:Pin1(W34A), the amount of co-immunoprecipitated WWP1 was significantly lower than that from H1299:Pin1(W34A); at the same time, immunoprecipitation with Myc antibody yielded similar amounts of either Myc-TAp63α(T538A) or WWP1 from H1299:Pin1 and H1299:Pin1(W34A).
These data suggest that Pin1 protects TAp63α from degradation via binding to its phospho-Thr538-Pro peptidyl-prolyl bonds and impairing the binding of WWP1 to the P540PxY543 motif of TAp63α; T538A mutation in TAp63α abolishes the action of Pin1 on this site and it makes TAp63α(T538A) proteins vulnerable to WWP1-mediated degradation.
Pin1 enhances TAp63α-mediated transcription and apoptosis
To investigate whether the Pin1-mediated upregulation of TAp63α can stimulate transcription of its target genes, luciferase reporter assays were performed. Our results demonstrate that TAp63α upregulated the Bax-luc reporter activities (Figure 5a), which was further enhanced by Pin1 (Figure 5b) in a dose-dependent manner. In addition, TAp63α stimulated the expression of endogenous Puma, which was further enhanced by Pin1 (Figures 5c and d). These results suggest that Pin1 has a positive role in regulating expression of downstream target genes of TAp63α, consistent with the results from a previous report.8

Pin1 enhances TAp63α-mediated transcription and cell apoptosis. (a) TAp63α upregulates the expression of Bax-luc reporter in a dose-dependent manner. H1299 cells were co-transfected with 500 ng of Bax-luc reporter, 50 ng pCMV-Renilla reporter and increasing amounts of TAp63α expression plasmid (0, 20, 50, and 100 ng). Twenty-four hours post-transfection, the cells were lysed and Bax-luc and Renilla activities were measured. Immunoblotting analysis was performed to detect the expression of TAp63α. The Bax-Luc activity was normalized to Renilla activity and presented as fold activation with standard deviation (n=3). (b) Pin1 upregulates TAp63α-mediated Bax-luc expression in a dose-dependent manner. H1299 cells were co-transfected with 500 ng of Bax-luc reporter, 50 ng pCMV-Renilla reporter, 20 ng TAp63α expression plasmid and an increasing amount of pcDNA3.1-HA-Pin1 (0, 100, 200, and 5 ng). Twenty-four hours post-transfection, luciferase and Renilla activities were measured. The Bax-Luc activity was normalized to Renilla activity and presented as fold activation with standard deviation (n=3). Immunoblotting analysis was performed to detect the expression of TAp63α and Pin1. (c) Pin1 upregulates TAp63α-mediated expression of endogenous Puma. H1299:Pin1 or H1299:Pin1(W34A) cells were transfected with TAp63α or its vector control (200 ng). Forty-eight hours post-transfection, cells were lysed and subjected to immunoblotting analysis. The intensities of Puma bands were normalized with Actin. (d) Pin1 upregulates TAp63α and endogenous Puma in a dose-dependent manner. H1299 cells were transfected with fixed dose of TAp63α plasmid (100 ng), together with an increasing amount of pcDNA3.1-HA-Pin1 (0, 200, 500 and 1000 ng). Forty-eight hours post-transfection, the cells were lysed and subjected to immunoblotting analysis with indicated antibodies. The intensities of Puma bands were normalized with Actin. (e) and (f) Pin1 augments TAp63α-induced cell apoptosis. TAp63α (400 ng) was co-transfected with HA-Pin1 or HA-Pin1(W34A) (1600 ng) into H1299 cells in a 6-well plate. Twenty-four hours after transfection, cells were trypsinized and subjected to immunoblotting analysis (e) or flow cytometry analysis of apoptosis (f and Supplementary Figure S4)
To examine the effect of Pin1 on TAp63α-mediated apoptosis, H1299 cells co-transfected with TAp63α and/or Pin1 were subjected to immunoblotting or flow cytometry (FCM) analysis. The immunoblotting results showed that TAp63α induced the cleavage of PARP1, which is a hallmark of apoptosis; wild-type Pin1, but not Pin1(W34A), further increases TAp63α-induced PARP1 cleavage (Figure 5e). FCM analysis further confirmed that wild-type Pin1, but not Pin1(W34A), promoted TAp63α-induced apoptosis (Figure 5f and Supplementary Figure S4).
Ablation of Pin1 decreases expression of ΔNp63α, leading to inhibition of cell proliferation
Our above-mentioned results demonstrated that, like TAp63α, ΔNp63α is also stabilized by Pin1 (Figures 2d, e, h, and 4d). To investigate whether the Pin1 effect on ΔNp63α regulates cell proliferation and tumorigenesis, we established stable FaDu cells with knockdown of Pin1 (shPin1), simultaneous knockdown of both Pin1 and WWP1 (shPin1/shWWP1), or overexpression of ΔNp63α in Pin1-knockdown cells (shPin1/ΔNp63α). As shown in Figure 6a, knockdown of Pin1 in FaDu cells decreased ΔNp63α (lane 2 versus lane 1), which was rescued by simultaneous knockdown of WWP1 (lane 3 versus lane 2). Notably, shPin1/ΔNp63α cells did not yield much higher protein levels of ΔNp63α than control cells (Figure 6a, lane 4 versus lane 1). This might be because Pin1 knockdown made both endogenous and exogenous ΔNp63α vulnerable to WWP1-mediated proteasomal degradation. Importantly, Pin1 knockdown led to significant decrease in cell proliferation, as shown by cell doubling time (Figure 6b) as well as FCM analysis (Figures 6c and d); simultaneous knockdown of WWP1 largely rescued proliferation defect induced by knockdown of Pin1 (Figures 6b–d). Furthermore, ectopic expression of ΔNp63α similar to knockdown of WWP1, was able to dramatically rescue inhibition of proliferation induced by Pin1 knockdown (Figures 6b–d). These data suggest that Pin1-mediated modulation of ΔNp63α has an important role in cell proliferation.

Pin1 knockdown inhibits cell proliferation via decreasing ΔNp63α protein levels. (a) Pin1 and WWP1 modulate the protein level of ΔNp63α in FaDu cells. FaDu cells were infected with shPin1, shWWP1 or ΔNp63α lentiviral particles sequentially, and screened with 2.5 μg/ml puromycin. Parts of cells were collected and lysed for immunoblotting analysis. (b) Pin1 knockdown induces inhibition of cell proliferation, which is rescued by simultaneous knockdown of WWP1 or overexpression of ΔNp63α. Stable FaDu cells mentioned above were seeded into 12-well plate at a density of 10 000 cells per well in triplicate. Cells were trypsinized and counted at indicated time points. (c) and (d) Pin1 knockdown induces G1 arrest of cell cycle, which is rescued by simultaneous knockdown of WWP1 or overexpression of ΔNp63α. Stable FaDu cells mentioned above were subjected to flow cytometry analysis (c), and percentages of cells in each phase were analyzed (d)
Pin1-mediated stabilization of ΔNp63α has important roles in tumorigenesis
To investigate whether Pin1-mediated stabilization of ΔNp63α affects cell proliferation in vivo, we monitored tumor formation derived from FaDu cells in nude mice. The results showed that control FaDu cells can develop observable tumors, while ablation of Pin1 (FaDu:shPin1) led to dramatically reduced tumor formation (Figures 7a–c). Importantly, simultaneous knockdown of both Pin1 and WWP1 (FaDu:shPin1/shWWP1) led to tumor formation much faster than FaDu:shPin1, although slower than controls (Figures 7a–c). In addition, ectopic expression of ΔNp63α could also rescue the tumor growth defect resulted from Pin1 knockdown (Figures 7a–c). These data suggest that Pin1 is critical for tumor formation in vivo, at least in part, through the WWP1-ΔNp63α pathway.

Pin1-mediated stabilization of ΔNp63α has important roles in tumorigenesis. (a)–(c) Pin1 knockdown induces growth inhibition of xenograft, which is rescued by simultaneous knockdown of WWP1 or ectopic expression of ΔNp63α. Stable FaDu cells mentioned above (2 × 106/0.1 ml) were subcutaneously inoculated into the right flank of each nude mice (n=5). The volumes of xenografts were measured 15, 20, 22, and 24 days after inoculation (a). The xenograft tumors dissected at day 24 were imaged (b) and weighed (c). **P<0.01. (d) Protein levels of ΔNp63α and Pin1 were correlated in oral squamous cell carcinoma (OSCC). Lysates of OSCC tumor tissues or paracancerous normal controls were subjected to immunoblotting analysis (shown as Supplementary Figure S5). Expression levels of ΔNp63α and Pin1 were normalized with actin, and ratio of either ΔNp63α or Pin1 in each paracancerous normal control was respectively set as 1.0 and indicated as the horizontal line
It has been shown that ΔNp63α is overexpressed in approximately 60% of oral squamous cell carcinoma (OSCC).14 To investigate whether Pin1-ΔNp63α pathway has a clinical relevance, we examined the protein expression by immunoblotting with OSCC tumor tissues and paracancerous normal controls. As shown in Figure 7d and Supplementary Figure S5, compared with their paracancerous normal controls (indicated as the horizontal line), protein levels of ΔNp63α and Pin1 were simultaneously elevated in 5 (1#, 3#, 6#, 8#, 12#) out of 12 OSCC patients. These data suggest that overexpression of Pin1 may have a role in stabilizing ΔNp63α in OSCC cells, that may contribute, in part, to the etiology of tumor development.
Discussion
p63 has been reported to have important roles in cell survival, proliferation, tumorigenesis, and development.2, 15 Even though accumulating evidence indicates that posttranslational regulation of p63 is important in control of p63 activity, how p63 protein stability is regulated remains still largely unclear. In this study, we show that peptidyl-prolyl isomerase Pin1 is an important regulator of p63. We found that Pin1 directly binds to p63 and disrupts p63α–WWP1 interaction, resulting in p63α protein stabilization. In addition, we show that Pin1 enhances TAp63α-mediated apoptosis and promotes ΔNp63α-mediated cell proliferation (as depicted in Figure 8). Furthermore, knockdown of Pin1 in FaDu cells inhibits tumor formation in nude mice, which is significantly rescued by simultaneous knockdown of WWP1 or ectopic expression of ΔNp63α. Moreover, overexpression of Pin1 correlates with increased expression of ΔNp63α in human oral squamous cell carcinoma samples.

Schematic model of a proposed mechanism showing how Pin1–p63α pathway modulates cell survival/proliferation. TAp63α and ΔNp63α undergo a proteasomal degradation mediated by E3 ligase WWP1. However, Pin1 may mediate the cis-trans isomerization or physical occupation of pTP-PPxY motifs of p63α proteins, leading to impairing their binding to WWP1. Consequently, TAp63α or ΔNp63α was stabilized. As a result, Pin1 inhibits cell survival/proliferation via stabilizing TAp63α or promotes cell survival/proliferation via stabilizing ΔNp63α
TAp63α contains 14 putative Pin1-binding sites, 8 of which are included in all of p63 proteins isoforms; there are 6 putative Pin1-binding sites in the unique C-terminal regions of α isoforms (as depicted in Figure 3a). Although the physical interaction between p63 and Pin1 was previously reported, the underlying mechanism and biological effects of Pin1–p63 direct interaction remain unclear.8 Our data show that Pin1 interacts with p63 proteins, including TAα, ΔNα, TAγ and ΔNγ isoforms. However, Pin1 specially stabilizes α isoforms, but not γ isoforms, indicating that these six Pin1-binding sites in the C-terminal region may have critical roles for protein stability of p63α isoforms. Although β isoforms have not been investigated in this work, we speculate that TAp63β and ΔNp63β can also be stabilized by Pin1, since they also contain the same TPPPxY sequence with p63α isoforms.
We further identified that T538 in TAp63α is critical for Pin1-mediated stabilization of p63α isoforms. Notably, T538 is adjacent to the WWP1 binding site (P540PxY543), and WWP1 has been reported to target its substrate proteins, including p63α, for proteasome-dependent degradation.12 Our data indicate that Pin1 inhibits the binding of WWP1 to p63α, likely because the isomerization resulted from the Pin1–p63 interaction affects the cyclization of the region encompassing PPxY motif of p63, which is important for the binding of WW domain of E3 ligases.16, 17 It is also possible that the binding of Pin1 to phospho-Thr538-Pro interferes with WWP1–p63α interaction, since both of them bind to p63 via their WW domains.12 Notably, since the E3 ligase Itch also binds to the same PPxY motif,16, 17, 18 it is plausible that Pin1 may also affect p63–Itch interaction. Moreover, it has been reported that ΔNp63α can also undergo a proteasomal degradation pathway coordinately mediated by MDM2 and Fbw7 in HaCaT and U2OS cells;19 it has been also documented that Pin1 can negatively regulate Fbw7 in human colon cancers and some cell lines including HCT116 and HeLa.20 Therefore, Pin1 may protect p63 proteins from proteasomal degradation mediated by MDM2/Fbw7. Our data suggest that Itch or MDM2/Fbw7 are unlikely to have major roles in Pin1-mediated p63α stabilization in our experiment system, since the decrease in p63α protein levels mediated by Pin1 knockdown is almost completely rescued by simultaneous knockdown of WWP1.
Though endogenous TAp63 proteins are barely detectable in somatic cells,21 overexpression of TAp63 proteins promotes cell cycle arrest and apoptotic cell death via activating p53 pro-apoptotic targets, such as Puma, Bax and Noxa.22, 23, 24 Animal studies reveal that TAp63 induces senescence and suppress tumorigenesis and metastasis in vivo.25, 26 In oocytes, TAp63α is responsible for DNA damage-induced apoptosis, therefore functioning as a quality control factor in the female germline.27, 28 Together, these studies indicate that TAp63 functions to promote apoptosis. In this study, we show that Pin1 enhances TAp63α protein stability, thereby stimulating TAp63α-mediated gene expression, including Puma, and leading to enhanced apoptosis.
In contrast to the tumor suppressor activity of TAp63, ΔNp63 proteins inhibit apoptosis and promote cell proliferation via antagonizing transcriptional activities of p53, TAp63 and TAp73 proteins.2 In the present work, we show that Pin1 reduces tumorigenicity of FaDu cells in xenograft animal models, indicating that Pin1 possesses strong oncogenic activity. FaDu is a human squamous carcinoma cell line, which expresses predominantly ΔNp63α. Our data indicate that knockdown of Pin1 leads to significant decrease in ΔNp63α protein level. Restoration of ΔNp63α protein levels, either by simultaneous knockdown of WWP1 or ectopic expression of ΔNp63α, significantly confers the tumor formation potential of the FaDu cells, indicating that the Pin1-WWP1-ΔNp63α pathway has important role in tumor development. Consistent with this notion, analysis of clinical samples reveals positive correlation of Pin1 and ΔNp63α expression in human oral squamous cell carcinoma. Although the amplification and transcription of p63 gene may contribute to p63 protein abundance in squamous cell carcinoma,29 Pin1 likely has some role in p63α stability, since both p63α and Pin1 proteins were readily detected in the clinical samples. Together, these studies suggest that Pin1-ΔNp63α pathway may be critical for development of human squamous carcinoma.
It was recently reported that Pin1 promotes mutant p53-dependent inhibition of the p63 function via potentiating the complex formed by Smad2, p63 and mutant p53 and downregulating p63 anti-metastatic target genes.8 In this work, we demonstrate that Pin1 can stabilize both TAp63α and ΔNp63α proteins, which functions as either a tumor suppressor or an oncoprotein, respectively (as depicted in Figure 8). Accumulating evidence indicates that Pin1 may exhibit tumor suppressor activity by positively regulating p53 and TAp73.9, 10, 11, 30 On the other hand, Pin1 is upregulated in human tumors and may promote tumorigenesis via activating oncoproteins such as mutant p53 or inactivating tumor suppressors such as Rb.8, 20, 31, 32, 33 In a given cell, the net effect of Pin1 may depend on the ratio of tumor suppressors (such as TAp63α) to oncoproteins (such as ΔNp63α) regulated by Pin1. Given the fact that ΔNp63α is the predominant p63 isoform and is overexpressed in human squamous carcinoma, our study suggests that Pin1 functions as an oncoprotein via activating ΔNp63α which may have important roles in certain tumor formation.
Materials and Methods
Constructs and siRNAs
Constructs of p63, Pin1 and Bax-luc reporters were previously described.34, 35, 36 The full-length human WWP1 cDNA was cloned into pCMV (Clontech, New York City, NY, USA). Pin1 shRNA sequence (TGCTGTTGACAGTGAGCGAAGGCCGAGTGTACTACTTCAATAGTGAAGCCACAGATGTATTGAAGTAGTACACTCGGCCTGTGCCTACTGCCTCGG) was cloned into pLKO.1 (Clontech). The construct of shWWP1 was purchased from Thermo Open Biosystem (Pittsburgh, PA, USA; target sequence: ATTGCTTATGAACGCGGCTTT).37 Small interference RNAs (siRNAs) were synthesized by GenePharma (Shanghai, China). The target sequences are as below: siWWP1, GCAGAGAAATACTGTTTAT;12 siPin1, GCCATTTGAAGACGCCTCG.38 The specific siRNAs or scrambled controls were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Cell culture, transfection and lentiviral infection
Cell culture and lentiviral infection were performed as described previously.34 Transient transfection was performed with Lipofectamin2000 (Invitrogen), and total amounts of plasmid DNA were balanced with corresponding vectors. H1299:Pin1, H1299:Pin1(W34A) and HEK:ΔNp63α cells were screened with 2 μg/ml puromycin. To generate FaDu:shPin1, FaDu:shPin1/shWWP1 and FaDu:shPin1/ΔNp63α cells, FaDu cells were infected with shPin1, shWWP1 or ΔNp63α lentiviral particles sequentially, and screened with 2 μg/ml puromycin.
For growth curve analysis, cells tranfected with indicated genes were trypsinized and reseeded into 12-well plate at a density of 10 000 cells per well in triplicate. In the following 3 days, cells were trypsinized and counted at indicated time points.
Pull-down, immunoprecipitation and immunoblotting analysis
GST-fused wild-type and mutant Pin1 proteins were expressed and subjected to pull-down assay as described previously.35 In brief, H1299 cells were transfected with expression plasmids of different p63 isoforms for 24 h and then lysed in EBC150 buffer containing 10% glycerol. A total of 500 μg of total proteins were subjected to GST pull-down assay with GST alone. The pull-down products or input samples were separated by 10% SDS-PAGE. Immunoblotting analysis (IB) was performed using indicated specific antibodies.
Immunoprecipitation (IP) and immunoblotting (IB) analysis were performed as previously described.11, 34 Antibodies used were specific for p63 (4A4 monoclonal antibody, Santa Cruz, Dallas, TX, USA, 1 : 200), myc (9E10 monoclonal antibody, Santa Cruz, 1 : 1000), Pin1 (rabbit polyclonal antibody, Cell Signaling, Danvers, MA, USA, 1 : 1000), actin (rabbit polyclonal antibody, Santa Cruz, 1 : 1000), HA (mouse monoclonal antibody, Millipore, Billerica, MA, USA, 1 : 500), WWP1 (rabbit monoclonal antibody, Epitomics, Burlingame, CA, USA, 1 : 3000), Puma (rabbit polyclonal antibody, Santa Cruz, 1 : 1000), and N-terminal cleaved PARP1 (rabbit polyclonal antibodies, Zenable, Chengdu, China, 1 : 8000).
Luciferase reporter, flow cytometry and protein stability assays
Luciferase reporter assay was performed with Dual-Luciferase Reporter (DLR) Assay System (Promega, Fitchburg, WI, USA) according to the manufacturer’s instructions. Flow cytometry (FCM) and protein stability assay was performed as described previously.34
Xenograft experiments
Male SCID mice in a BALB/C background, at an age of 8–10 weeks at the time of inoculation, were used to establish xenografts. Cells (2 × 106/0.1 ml) were subcutaneously inoculated into the right flank of each nude mice. The volumes of xenograft were measured 15, 20, 22, and 24 days after inoculation. The tumor volumes were calculated according to the formula:volume=length × width2. The xenografts were dissected and weights were measured 24 days after inoculation.
Human oral squamous cell carcinoma samples
Human specimens of oral squamous cell carcinoma and paracancerous tissues were dissected from patients at surgery. Tissues were stored in liquid nitrogen until processing. For immunoblotting analysis, the tissues were ground in liquid nitrogen and lysed with radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-Cl pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, plus 5 mM EDTA, 1 mM NaF, 1 mM sodium orthovanadate, and protease inhibitors).
Quantification and statistical analysis
Intensities of protein bands were quantified with Image Lab (Bio-Rad, Berkeley, CA, USA) and relative intensities were normalized with actin. Data were shown as means±standard deviations (S.D.). P-values were calculated using the Student’s t test.
Abbreviations
- TAα:
-
TAp63α
- TAβ:
-
TAp63β
- TAγ:
-
TAp63γ
- ΔNα:
-
ΔNp63α
- ΔNβ:
-
ΔNp63β
- ΔNγ:
-
ΔNp63γ
- WT:
-
wild-type
- T or Thr:
-
threonine
- S or Ser:
-
serine
- P or Pro:
-
proline
- A or Ala:
-
alanine
- W or Trp:
-
tryptophan
- Y or Tyr:
-
tyrosine
- CHX:
-
cycloheximide
- IP:
-
immunoprecipitation
- IB:
-
immunoblotting
References
Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 1998; 2: 305–316.
Murray-Zmijewski F, Lane DP, Bourdon JC . p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ 2006; 13: 962–972.
Lu KP, Liou YC, Zhou XZ . Pinning down proline-directed phosphorylation signaling. Trends Cell Biol 2002; 12: 164–172.
Shaw PE . Peptidyl-prolyl isomerases: a new twist to transcription. EMBO Rep 2002; 3: 521–526.
Jin H, Jiang J, Sun L, Zheng F, Wu C, Peng L et al. The prolyl isomerase Pin1 is overexpressed in human esophageal cancer. Oncol Lett 2011; 2: 1191–1196.
Chen CH, Chang CC, Lee TH, Luo ML, Huang P, Liao PH et al. SENP1 desumoylates and regulates Pin1 protein activity and cellular function. Cancer Res 2013; 73: 3951–3962.
Liou YC, Zhou XZ, Lu KP . Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem Sci 2011; 36: 501–514.
Girardini JE, Napoli M, Piazza S, Rustighi A, Marotta C, Radaelli E et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011; 20: 79–91.
Mantovani F, Piazza S, Gostissa M, Strano S, Zacchi P, Mantovani R et al. Pin1 links the activities of c-Abl and p300 in regulating p73 function. Mol Cell 2004; 14: 625–636.
Zacchi P, Gostissa M, Uchida T, Salvagno C, Avolio F, Volinia S et al. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 2002; 419: 853–857.
Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf G et al. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 2002; 419: 849–853.
Li Y, Zhou Z, Chen C . WW domain-containing E3 ubiquitin protein ligase 1 targets p63 transcription factor for ubiquitin-mediated proteasomal degradation and regulates apoptosis. Cell Death Differ 2008; 15: 1941–1951.
Stewart D, Kazemi S, Li S, Massimi P, Banks L, Koromilas AE et al. Ubiquitination and proteasome degradation of the E6 proteins of human papillomavirus types 11 and 18. J Gen Virol 2004; 85 (Pt 6): 1419–1426.
Lo Muzio L, Campisi G, Farina A, Rubini C, Pastore L, Giannone N et al. Effect of p63 expression on survival in oral squamous cell carcinoma. Cancer Invest 2007; 25: 464–469.
Dotsch V, Bernassola F, Coutandin D, Candi E, Melino G . p63 and p73, the ancestors of p53. Cold Spring Harb Perspect Biol 2010; 2: a004887.
Bellomaria A, Barbato G, Melino G, Paci M, Melino S . Recognition mechanism of p63 by the E3 ligase Itch: novel strategy in the study and inhibition of this interaction. Cell Cycle 2012; 11: 3638–3648.
Bellomaria A, Barbato G, Melino G, Paci M, Melino S . Recognition of p63 by the E3 ligase ITCH: Effect of an ectodermal dysplasia mutant. Cell Cycle 2010; 9: 3730–3739.
Rossi M, Aqeilan RI, Neale M, Candi E, Salomoni P, Knight RA et al. The E3 ubiquitin ligase Itch controls the protein stability of p63. Proc Natl Acad Sci USA 2006; 103: 12753–12758.
Galli F, Rossi M, D’Alessandra Y, De Simone M, Lopardo T, Haupt Y et al. MDM2 and Fbw7 cooperate to induce p63 protein degradation following DNA damage and cell differentiation. J Cell Sci 2010; 123 (Pt 14): 2423–2433.
Min SH, Lau AW, Lee TH, Inuzuka H, Wei S, Huang P et al. Negative regulation of the stability and tumor suppressor function of Fbw7 by the Pin1 prolyl isomerase. Mol Cell 2012; 46: 771–783.
Paris M, Rouleau M, Puceat M, Aberdam D . Regulation of skin aging and heart development by TAp63. Cell Death Differ 2012; 19: 186–193.
Osada M, Ohba M, Kawahara C, Ishioka C, Kanamaru R, Katoh I et al. Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat Med 1998; 4: 839–843.
Wu G, Nomoto S, Hoque MO, Dracheva T, Osada M, Lee CC et al. DeltaNp63alpha and TAp63alpha regulate transcription of genes with distinct biological functions in cancer and development. Cancer Res 2003; 63: 2351–2357.
Gressner O, Schilling T, Lorenz K, Schulze Schleithoff E, Koch A, Schulze-Bergkamen H et al. TAp63alpha induces apoptosis by activating signaling via death receptors and mitochondria. EMBO J 2005; 24: 2458–2471.
Guo X, Keyes WM, Papazoglu C, Zuber J, Li W, Lowe SW et al. TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat Cell Biol 2009; 11: 1451–1457.
Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin YL et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 2010; 467: 986–990.
Amelio I, Grespi F, Annicchiarico-Petruzzelli M, Melino G . p63 the guardian of human reproduction. Cell Cycle 2012; 11: 4545–4551.
Gonfloni S, Di Tella L, Caldarola S, Cannata SM, Klinger FG, Di Bartolomeo C et al. Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nat Med 2009; 15: 1179–1185.
Hibi K, Trink B, Patturajan M, Westra WH, Caballero OL, Hill DE et al. AIS is an oncogene amplified in squamous cell carcinoma. Proc Natl Acad Sci USA 2000; 97: 5462–5467.
Teng BL, Hacker KE, Chen S, Means AR, Rathmell WK . Tumor suppressive activity of prolyl isomerase Pin1 in renal cell carcinoma. Mol Oncol 2011; 5: 465–474.
Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V et al. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J 2001; 20: 3459–3472.
Wulf G, Ryo A, Liou YC, Lu KP . The prolyl isomerase Pin1 in breast development and cancer. Breast Cancer Res 2003; 5: 76–82.
Rizzolio F, Lucchetti C, Caligiuri I, Marchesi I, Caputo M, Klein-Szanto AJ et al. Retinoblastoma tumor-suppressor protein phosphorylation and inactivation depend on direct interaction with Pin1. Cell Death Differ 2012; 19: 1152–1161.
Li X, Chen J, Yi Y, Li C, Zhang Y . DNA damage down-regulates DeltaNp63alpha and induces apoptosis independent of wild type p53. Biochem Biophys Res Commun 2012; 423: 338–343.
Sdek P, Ying H, Chang DL, Qiu W, Zheng H, Touitou R et al. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol Cell 2005; 20: 699–708.
Ying H, Chang DL, Zheng H, McKeon F, Xiao ZX . DNA-binding and transactivation activities are essential for TAp63 protein degradation. Mol Cell Biol 2005; 25: 6154–6164.
Subik K, Shu L, Wu C, Liang Q, Hicks D, Boyce B et al. The ubiquitin E3 ligase WWP1 decreases CXCL12-mediated MDA231 breast cancer cell migration and bone metastasis. Bone 2012; 50: 813–823.
Rustighi A, Tiberi L, Soldano A, Napoli M, Nuciforo P, Rosato A et al. The prolyl-isomerase Pin1 is a Notch1 target that enhances Notch1 activation in cancer. Nat Cell Biol 2009; 11: 133–142.
Acknowledgements
This work was supported by National Basic Research Program of China (#2012CB910700 to ZX Xiao) and National Natural Science Foundation of China (#31100982 to C Li and #31171362 to ZX Xiao). We thank Han Kang and Anning Han for technical support, Dr Yujun Zhang and Dr Richard Eckner (University of Medicine and Dentistry of New Jersey) for helpful discussions.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Li, C., Chang, D., Yang, Z. et al. Pin1 modulates p63α protein stability in regulation of cell survival, proliferation and tumor formation. Cell Death Dis 4, e943 (2013). https://doi.org/10.1038/cddis.2013.468
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.468
Keywords
This article is cited by
-
WWP1 E3 ligase at the crossroads of health and disease
Cell Death & Disease (2023)
-
The emerging role of WWP1 in cancer development and progression
Cell Death Discovery (2021)
-
TP63 links chromatin remodeling and enhancer reprogramming to epidermal differentiation and squamous cell carcinoma development
Cellular and Molecular Life Sciences (2020)
-
ΔNp63α down-regulates c-Myc modulator MM1 via E3 ligase HERC3 in the regulation of cell senescence
Cell Death & Differentiation (2018)
-
Prolyl isomerase Pin1: a promoter of cancer and a target for therapy
Cell Death & Disease (2018)
