
Abstract
Retinoic acid (RA) contributes to cleft palate; however, the cellular and molecular mechanisms responsible for the deleterious effects on the developing palate are unclear. Wnt signaling is a candidate pathway in the cleft palate and is associated with RA in organ development; thus, we aim to investigate whether RA-induced cleft palate also results from altered Wnt signaling. Administration of RA to mice altered cell proliferation and apoptosis in craniofacial tissues by regulating molecules controlling cell cycle and p38 MAPK signaling, respectively. This altered cell fate by RA is a crucial mechanism contributing to 100% incidence of cleft palate. Moreover, Wnt/β-catenin signaling was completely inhibited by RA in the early developing palate via its binding and activation with RA receptor (RAR) and is responsible for RA-induced cleft palate. Furthermore, PI3K/Akt signaling was also involved in actions of RA. Our findings help in elucidating the mechanisms of RA-induced cleft palate.
Similar content being viewed by others

Main
The whole process of palatal development is precisely regulated. Disruption at any stage of growth – elevation or fusion, genetically or environmentally – causes cleft palate. However, the underlying developmental pathogenesis remains unclear.
Palatal shelves, composed of a core of mesenchyme covered by an epithelium, grow vertically on both sides of the tongue from embryonic day 13.5 (E13.5) in the mouse. Concomitant with the enlarging lower jaw and lowering tongue at E14.5, the shelves elevate to a horizontal position above the dorsum of the tongue. At about E15.5, the medial edge epithelium (MEE) of the horizontal palatal shelves contact, adhere and fuse along their midline, forming a midline epithelial seam. At E17.5, the continuous palate formation separates the oral and nasal cavities for breathing and feeding at the same time.
Retinoic acid (RA) has an essential role during embryonic development of regulating morphogenesis, cell proliferation and differentiation, and extracellular matrix production.1, 2, 3 RA is widely used for skin diseases; however, excess exogenous RA can adversely affect craniofacial development in rodents and humans.4, 5 Cleft palate is one of the major malformations induced by RA in mouse fetuses. RA may have a direct effect on the fetus, disturb polysaccharide metabolism of the ground substance of the palatal processes, disturb the blood concentration of thyroid hormone and interfere with carbohydrate metabolism.4, 6 Notably in recent years, RA was linked to Wnt (wingless) signaling in organ development and disease therapy.7, 8, 9 However, the mechanisms of action of RA in cleft palate formation are still not fully understood.
Although not yet implicated by genome-wide association studies, variants within WNT genes were found to be associated with cleft lip and palate,10 and mutations in WNT3 underlie autosomal-recessive tetra-amelia with cleft lip and palate.11 These findings have led to further analyses of genes in the WNT signaling pathway as candidates for normal development of the lip and the palate. Wnt3A, Wnt5A and Wnt11 were significantly associated with non-syndromic cleft lip with or without cleft palate.10 The Wnt molecules act through specific receptors to activate intracellular signals that dictate a variety of cell actions, including proliferation, migration, growth, differentiation and apoptosis.
We hypothesized that RA-induced cleft palate was associated with Wnt signaling by altering palatal cell fate in the crucial time of craniofacial development. To verify our assumption, we analyzed the protein expression of Wnt signaling molecules and investigated cell proliferation and survival after RA exposure from E13.5 to E15.5.
Results
Embryos exposed to RA showed 100% incidence of the cleft palate
We chose 100 mg/kg RA at E12 according to previous works5, 14 and found survival of few litters and all viable embryos with cleft palate and severe limb abnormalities. Thus, we used 70 mg/kg RA15, 16 at E12. Consistent with our expectation, the 100% of fetuses showed cleft palate with litter death and few other malformations (Supplementary Table S1). No controls showed cleft palate. RA-treated E17.5 embryos showed a wider, complete secondary cleft palate than did controls (Supplementary Figure S1) because of unelevated palate, which was manifested by our morphology results of RA-treated embryos.
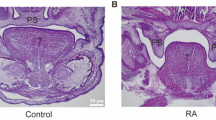
At E13.5, interestingly, RA-exposed embryos showed no grooves between the palatal shelf and the body of maxilla in the anterior palatal shelves (Figure 1a, arrow heads); however, grooves were observed in the controls (Figure 1a, arrows). The posterior vertical growth of palatal shelves did not differ between RA-treated embryos (Figure 1a) and controls (Figure 1a). E14.5 embryos of controls, even from the same litter, showed various anterior and posterior shelf morphologies, from vertically oriented (Figure 1b) to bilaterally elevated (Figure 1b). However, all E14.5, RA-exposed embryos showed vertically oriented anterior and posterior shelves (Figure 1b). Notably, the grooves between the palatal shelf and the body of maxilla were not observed in the anterior palatal shelves in RA-treated embryos (Figure 1b, arrow heads) as compared with unelevated controls (Figure 1b, arrows) at E14.5. As compared with the open space of the oral–nasal cavity in the posterior portions between control E13.5 (Figure 1a, bipitch arrow) and E14.5 embryos (Figure 1b, bipitch arrow) and between RA-exposed E14.5 (Figure 1b, bipitch arrow) and control E14.5 embryos (Figure 1b, bipitch arrow), the space for E14.5 controls was larger than others. However, RA-exposed E13.5 (Figure 1a, bipitch arrow) and E14.5 embryos (Figure 1b, bipitch arrow) did not differ in the space. All control shelves had completely contacted and adhered (Figure 1c) by E15.5, whereas the anterior and posterior palatal shelves of RA-treated embryos were still unelevated (Figure 1c). This condition was observed at E17.5; as compared with the complete secondary palate formation for controls (Figure 1d), a large gap eventually manifested as a wider cleft phenotype with RA treatment (Figure 1d).

Palate development in RA-treated embryos. E13.5 ( a), E14.5 (b), E15.5 (c) and E17.5 (d) of controls and RA-treated embryos showing histological frontal sections in anterior and posterior (at the level of the first lower molar tooth) regions of developing palates. Arrows for controls and arrow heads for RA-exposed embryos point to the grooves between the palatal shelf and the body of maxilla. Bipitch arrows point to the open space of oral–nasal cavity. Con, control; ps, palatal shelf; t, tongue. Bars, (a and b, c except RA-exposed sections) 200 μm and (c except Control sections and d) 250 μm
Anterior palatal mesenchyme proliferation was significantly increased in RA-treated embryos
As RA treatment greatly downregulated Wnt/β-catenin signaling in developing craniofacial tissues at E13.5 and E14.5 and this pathway has a crucial role in cell fate such as cell proliferation, differentiation, adhesion and survival, we examined cell turnover by BrdU incorporation (Figure 2, red fluorescence) and TUNEL staining (Figures 3 and 4, green fluorescence). During E13.5–E15.5 (Figures 2a–d), BrdU-positive cells in the mesenchyme of the right (Figure 2d, columns 1 and 3 to P=0.01 and 0.038, respectively) and left (Figure 2d, columns 1, 2 and 3 to P=0.045, 0.027 and 0.012, respectively) anterior palatal shelves were higher for RA-treated embryos than controls. However, BrdU-labeled cells in the mesenchyme of the posterior palatal shelves with RA treatment did not differ from those of controls from E13.5 to E15.5 (Figures 2a–d). In contrast, the epithelium proliferation of palatal shelves was not altered with RA as compared with control treatment from E13.5 to E15.5 (Figures 2a–d and Supplementary Figure S2).

Cell proliferation in palatal shelves with RA treatment. Immunostaining for incorporated BrdU (red fluorescence) in developing palates from the anterior and posterior portions of controls and RA-treated embryos from E13.5 to E15.5 (a–c). White lines demarcate the palatal region for counting of BrdU-labeled cells in the epithelium and mesenchyme. DAPI-stained cell nuclei (blue fluorescence). Arrows point to the grooves between the palatal shelf and the body of maxilla in controls. Quantification of proportion of BrdU-positive cells in the mesenchyme (d) of fixed area in palatal shelves of controls and RA-treated embryos from E13.5 to E15.5. Data are mean±S.D. *P<0.05. Con, control; M, mesenchyme; mee, medial edge epithelium; nse, nasal side epithelium; ose, oral-side epithelium; ps, palate shelf; t, tongue. Bars, 50 μm. (e) Protein levels of p27, p21 and cyclin D1 before and after RA treatment. Primary MEPM was treated with 2 μM RA for indicated time

Cell survival in palatal shelves with RA treatment. TUNEL-positive cells (green fluorescence) in anterior regions of controls (a for E13.5, white boxes; b for E14.5, arrows) and RA-treated palatal shelves (a for E13.5, white triangles; b for E14.5, arrow heads). At E15.5, TUNEL-positive cells in the remainders of midline epithelial seam of fused palate of controls and the medial edge epithelium of RA-treated unelevated palatal shelves (c, asterisks). DAPI-stained cell nuclei (blue fluorescence). Con, control; ps, palatal shelf; t, tongue. Bars, 50 μm

Altered TUNEL-positive cells in RA-treated embryos. At E13.5, TUNEL-positive cells (green fluorescence) in the bend between the palatal shelf and the cranial base with RA treatment (a, arrow head) and controls (a, arrow). TUNEL-positive cells in the floor of mouth epithelium of the posterior regions of controls (b, white boxes) and of RA-exposed embryos (b, white triangles) at E13.5. Clustered TUNEL-positive cells in the anterior mesenchyme of the primordial frenulum (c, yellow asterisk) and posterior mesenchyme of the primordial genioglossus muscle (c, orange asterisk) of RA-treated embryos and controls (c, white asterisk) at E13.5. At E14.5, extensive TUNEL-positive cells were found in the mesenchyme of the primordial frenulum with RA treatment (d, orange box) and controls (d, white triangles). DAPI-stained cell nuclei (blue fluorescence). Con, control; f, floor of mouth; ps, palatal shelf; t, tongue. Bars, 50 μm. (e) Protein level of phospho-p38 MAPK before and after RA treatment. Primary MEPM cells were treated with 2 μM RA for indicated time. (f) Inhibition of p38 MAPK rescued RA-decreased cell viability in MEPM cells. Cells were pretreated with 10 μM SB202190 for 1 h, followed by 2 μM RA treatment for 24 h. Data are shown as mean±S.E. (n=6). ***P<0.001. (g) Inhibition of p38 MAPK prevented RA-increased caspase-3 activity in MEPM cells. The treatments were the same as in f. Data are shown as mean±S.E. (n=6). **P<0.01
To further clarify the signaling pathway involved in RA-induced proliferation, an in vitro primary MEPM cell model was used to detect the effects of RA on the expression of p21, p27 and cyclin D1, all of which are key regulators to cell cycle. Interestingly, the protein levels of p21 and p27, two negative cell cycle regulators, increased in a time-dependent manner within 24 h, whereas decreased after a longer treatment (48 h; Figure 2e). Consistently, the expression of cyclin D1, a positive cell cycle regulator, exhibited a reverse trend (Figure 2e). This result indicates that the RA-induced cell proliferation in vivo may be mainly ascribed to the RA-changed cell cycle.
Apoptosis of the epithelium and mesenchyme was altered by RA at E13.5 and E14.5
At E13.5, clustered TUNEL-positive cells were detected in the oral-side epithelium of anterior regions of palatal shelves in controls (Figure 3a, white boxes) but not in RA-treated embryos (Figure 3a, white triangles). Moreover, the amount of TUNEL-positive cells in the mesenchyme of the bend between the palatal shelf and the cranial base was greater for RA-treated embryos (Figure 4a, arrow head) than the controls (Figure 4a, arrow); and the amount of TUNEL-labeled cells in the epithelium of the floor of mouth was lower for RA-treated embryos (Figure 4b, white triangles) than controls (Figure 4b, white boxes) at E13.5. A substantial amount of TUNEL-positive cells was detected in the anterior mesenchyme of the primordial frenulum (Figure 4c, yellow asterisk) and posterior mesenchyme of the primordial genioglossus muscle (Figure 4c, orange asterisks) with RA than control treatment (Figure 4c, white asterisk). At E14.5, apoptotic cells were evident in the oral-side epithelium of the anterior portions of palatal shelves for controls (Figure 3b, arrows) but not for RA-treated embryos (Figure 3b, arrow heads). Interestingly, we detected apoptotic cells in the epithelium of the floor of the mouth for controls (Figure 4d, white triangles) but not for RA-treated embryos (Figure 4d, orange triangles). However, we detected clustered TUNEL-labeled cells in the mesenchyme of the primordial frenulum of RA-treated embryos (Figure 4d, orange box) but not controls (Figure 4d, white box). Surprisingly, at E15.5, TUNEL-positive cells were detected in the anterior MEE of palatal shelves with RA treatment and control treatment (Figure 3c, asterisks).
It has been reported that the p38 MAPK signaling pathway was highly involved in RA-induced apoptotic cell death.17, 18 To further determine this issue in our model, we treated primary MEPM cells with RA. Western blot showed a time-dependent stimulatory effect of RA on the protein level of phospho-p38 MAPK (T180/T182) within 24 h; however, these effects decreased after a longer treatment (48 h; Figure 4e). In line with RA-activated p38 MAPK, RA significantly decreased the cell viability of primary MEPM cells, which was blocked by SB202190, a specific p38 MAPK inhibitor (Figure 4f). Similarly, SB202190 also prevented RA-increased caspase-3 activity (Figure 4g). These results together suggest that p38 MAPK is a key effector in RA-induced cell apoptosis.
Wnt/β-catenin signaling was inhibited by RA in E13.5 and E14.5
Wnt signaling pathway proteins were detected in developing mouse palatal shelves and tongue extracts of RA-treated embryos and controls from E13.5 to E15.5 (Figure 5a). Interestingly, in E13.5 and E14.5, Gsk-3β expression showed a downward shift in RA-treated embryos as compared with controls, which indicated the sharply reduced phospho-Gsk-3β expression and increased activity of Gsk-3β. Consistent with the altered pospho-Gsk-3β level, the downstream molecules of Wnt signaling, including β-catenin, Lef-1 and c-Jun and phospho-c-Jun, were completely downregulated in E13.5 and E14.5 embryos (Figure 5a). However, the level of pospho-Gsk-3β did not differ from that of controls at E15.5. Consistently, the downstream molecules of Wnt signaling, β-catenin and Lef-1 were slightly upregulated with RA at E15.5, with no difference in the protein level of c-Jun and phospho-c-Jun between RA samples and controls (Figure 5a). Notably, levels of Wnt3a, Wnt5a and Wnt11 – Wnt ligands – were not markedly different from those of the controls (Figure 5a). These results together demonstrate that the Wnt/β-catenin signaling pathway was targeted and inhibited by RA. To further verify whether the downregulation of Wnt signaling results from a direct RA effect, IWR-1-endo was used to block Wnt/β-catenin signaling in primary MEPM cells. We found that IWR highly mimicked the RA’s effect of reducing cell viability (Figure 5b). Interestingly, a synergism was observed for the use of IWR in combination with RA, compared with IWR alone (Figure 5b). These data demonstrate the central role of the Wnt/β-catenin signaling pathway in RA-induced palate deformation, although other secondary signaling pathways may also be targeted.

RA inhibited Wnt/β-catenin signaling in E13.5 and E14.5. (a) Western blot analysis of protein levels in mouse palatal shelves and tongue extracts with RA and control treatment at E13.5, E14.5 and E15.5. Samples were run on the same gel and were contiguous. (b) Inhibition of Wnt signaling mimicked the effect of RA. MEPM cells were pretreated with 10 μM IWR-1-endo for 4 h, followed by 2 μM RA treatment for 24 h. Data are shown as mean±S.E. (n=6). ***P<0.001. IWR, IWR-1-endo
RA actions were mediated by its binding and activation of RARs
The effects of RA in vertebrates are mediated by its binding and activation of two families of nuclear receptors, RAR and RXR.19 These receptors bind to RA response elements leading to activation of many signaling pathways. To clarify the roles of these two receptors, we treated primary MEPM cells with a serial concentration of RA, and the mRNA levels of RAR and RXR were analyzed using quantitive real-time PCR. As shown in Figure 6a, RA treatment for 24 h caused the activation of RARβ and RARγ in a dose-response manner, but not of RARα (Figure 6a). Nevertheless, RA showed weak stimulatory effects on RXR, of which only RXRγ exhibited a relative better RA-response sensitivity (Figure 6b). To further confirm the role of RAR on the actions of RA, the effect of BMS493, a pan RA receptor antagonist, was studied on RA-treated MEPM cells. Pretreatment with BMS493 significantly rescued RA-suppressed cell viability (Figure 6c) and depressed RA-elevated caspase-3 activity (Figure 6d). These data clearly indicate that RA actions are dependent on RAR.

RAR is required for the actions of RA. (a) Real-time quantitative PCR was used to quantify the mRNA level of RAR in MEPM cells treated with various concentrations of RA for 24 h. The level of RAR was normalized to β-action. Data are expressed as the mean±S.E. (b) Effect of RA on the mRNA level of retinoid X receptor. The treatments were the same as in a. Data are expressed as the mean±S.E. (c) Inhibition of RAR abolished RA-decreased cell viability. MEPM cells were pre-incubated for 20 min with or without 3 μM BMS493 and then treated with or without 2 μM RA for 24 h. Data are shown as mean±S.E. (n=3). ***P<0.001. (d) Inhibition of RAR blocked RA-increased caspase-3 activity. The treatments were the same as in c. Data are shown as mean±S.E. (n=3). **P<0.01. (e) Protein levels of Fgf10 and Shh before and after RA treatment. Primary MEPM cells were treated with 2 μM RA for indicated time. (f) PI3K/Akt signaling was involved in the actions of RA. Primary MEPM cells were treated with 2 μM RA alone for indicated time, or co-incubated with 5 μM LY294002 for 48 h. LY, LY294002
Discussion
The phenotype of RA-induced cleft palate was a wider gap, with palatal shelves still vertically growing on both sides of the tongue. Shelf development is achieved by distinct mechanisms in different regions along the anteroposterior axis,20 and clinical observations suggest that left and right shelf development, such as unilateral cleft lip or palate, is controlled by different mechanisms. Therefore, we analyzed morphological change in anterior and posterior regions with control and RA treatment of embryos. RA permanently impeded palatal shelf elevation, the main reason for the 100% incidence of cleft palate. RA totally changed the morphogenesis of craniofacial tissues in at least three aspects: (1) palatal shelves abnormally retained the capability of vertical growth from E13.5 to E15.5; (2) anterior palatal shelves lost the grooves between the palatal shelf and the body of maxilla at E13.5 and E14.5, which might obstruct the elevation of palatal shelves; and (3) the action of tongue withdrawal could not prepare enough space for elevation of palatal shelves.
Cell proliferation appeared in all craniofacial tissues, including the epithelium and mesenchyme, for a hyperproliferative situation in early embryo development but gradual reduction from E13.5 to E15.5. However, cell apoptosis appeared only at a specific location and time and was required for craniofacial development. The proliferation of palatal epithelium was not influenced by RA exposure; however, the apoptosis of the specific epithelium of RA-treated embryos was completely disordered in E13.5 and E14.5, which is consistent with the time of downregulated Wnt/β-catenin signaling. Thus, the apoptosis mechanism of oral-side epithelium of palatal shelves was important for elevation of palatal shelves and, if lost, does not permit the flip-up of palatal shelves. Moreover, the apoptosis mechanism of the epithelium of the floor of mouth helped in the descent of the tongue. The anterior palatal mesenchyme proliferation of RA-treated embryos was significantly increased from E13.5 to E15.5, and the posterior palatal mesenchyme proliferation increased with RA exposure from E13.5 to E15.5 but not significantly. Thus, increased proliferation of palatal mesenchyme allowed for continual growth of palatal shelves, which in turn may have disastrous effects on the subsequent palatal elevation and fusion. The specific mesenchyme apoptosis was increased by RA exposure at E13.5 and E14.5, which influenced the movement of the tongue. Interestingly, the mechanism of MEE apoptosis was indistinguishable from that of controls at E15.5; therefore, MEE apoptosis may not depend on contact between the palatal shelves but instead is temporally programmed.21
We found that RA completely inhibited the canonical Wnt/β-catenin signaling pathway at E13.5 and E14.5. By E15.5, β-catenin and Lef-1, upstream of Wnt/β-catenin signaling, were slightly upregulated as compared with controls. By using IWR-1-endo in primary MEPM cell model, we confirmed that Wnt/β-catenin signaling pathway is, although not the only, a very important target of RA because blocking Wnt/β-catenin signaling almost completely replicated the action of RA. Next, we demonstrated that RAR is required for RA to exert its biological actions. Thus, an RA-RAR-Wnt/β-catenin signaling axis is identified. Some previous studies demonstrated that Shh signaling from the oral region of the palatal epithelium is required for palatal mesenchymal proliferation,22, 23 and increased Shh signaling has a stimulatory role on cell proliferation in the palatal mesenchyme. Importantly, Fgf10 has restricted expression in the anterior palatal mesenchyme, regulating Shh expression, and cell proliferation and apoptosis in the palatal epithelium.24, 25 Several studies demonstrated a regulatory loop between Shh and Wnt signaling and that Shh is a downstream target of Wnt signaling and acts as a negative feedback regulator of Wnt signaling.26, 27, 28 A recent study showed that compromised canonical Wnt/β-catenin signaling reduced Fgf family gene expression and Fgf signaling activity.20 Therefore, it is speculated that RA downregulated Wnt/β-catenin signaling at E13.5 and E14.5, which in turn may also regulate Fgf and Shh signaling, thus contributing to altered palatal mesenchyme proliferation and apoptosis (Figure 7). Expectedly, we found that the protein level of Fgf10 and Shh decreased firstly and then increased after the RA treatment in vitro (Figure 6e), consistent with the existence of negative feedback loop. In addition, the PI3K/Akt signaling, of which one downstream mTORC1 controlling cell proliferation, whereas another mTORC2 controlling cell survival,29 also exhibited a similar change (Figure 6f), and its regulation by RA also explained the cell fate changed by RA. At E15.5, the downregulated Wnt/β-catenin signaling was not as apparent as at E13.5 and E14.5 mainly because of the negative feedback regulation of Shh on Wnt/β-catenin signaling (Figures 5a, 6e and 7). Taken together, it is reasonable to conclude that the proliferation and apoptosis of epithelium and mesenchyme are regulated by signaling molecules induced by RA exposure in the developing palate and finally contribute to the cleft palate (Figure 7).

Possible molecular cross-talk after RA exposure in developing palate. Scheme depicts in craniofacial tissues the interaction between canonical Wnt/β-catenin signaling and possible downstream signaling to control cell fate from E13.5 to E15.5. Arrows represent inductive relationships; flat ends indicate possible inhibitory relationships. Arrows following molecules or cell fate indicate up- or downregulation and numbers of arrows represent the degree of regulation. Dash line arrows represents possible downstream molecule. Equal signs indicate no difference of protein expression. Broken lines represent cell membrane; solid lines indicate nuclear membrane
In summary, administration of RA at E12 of mouse embryos altered the proliferation and apoptosis of palatal epithelium and mesenchyme and impeded the elevation of palatal shelves, thus leading to cleft palate. Our study identified a previously unknown molecular mechanism that the downregulation of canonical Wnt/β-catenin signaling is responsible for RA-induced cleft palate and that the cell fate altered by molecules controlling cell cycle or apoptosis directly shaped the morphologic features of RA-induced cleft palate. These findings are helpful for highlighting the molecular and the teratogenic actions of RA.
Materials and Methods
Animals
All experiments involving animals were approved by the Nan Fang Hospital Institutional Animal Care and Use Committee. Females of Kun Ming mice were crossed with fertile males overnight. Observation of a vaginal plug was designated as E0.5. Pregnant females at E12 were randomly divided into two groups for treatment: RA (Sigma, St. Louis, MO, USA) dissolved in sesame oil by gavage at 70 mg/kg, or 10 ml/kg sesame oil only as a control.
Scanning electron microscopy
Trimmed fetuses at E17.5 were fixed with 2.5% glutaraldehyde at 4 °C for 12 h. After dehydration with a graded acetone series, specimens were dried with liquid CO2 and coated with a thin layer of gold before being viewed under an S-3000 N scanning electron microscope.
Histochemical staining
Embryonic heads were fixed in 4% paraformaldehyde and dehydrated through an ethanol series and embedded in paraffin for sectioning by routine procedures. For general morphology, deparaffinized sections (5 μm) were stained with hematoxylin and eosin by standard procedures.
Western blot analysis
The palatal shelves and tongue extracts of control and RA-treated embryos from E13.5 to E15.5 were analyzed by standard protocols with antibodies for Wnt3a, Wnt5a and Wnt11 (all R&D Systems, Minneapolis, MN, USA); Gsk-3β, β-catenin, Lef-1, p27, Cyclin D1, phospho-p38 MAPK (T180/T182) and Shh (sonic hedgehog; all Cell Signaling Technology, Beverly, MA, USA); c-Jun, phospho-c-Jun (S63), p21 and FGF10 (fibroblast growth factor 10; all Epitomics, Hangzhou, China); and α-Tubulin, Akt and phospho-Akt (S473; Santa Cruz Biotechnology, Santa Cruz, CA, USA).
BrdU analysis and TUNEL
Pregnant females from E13.5 to E15.5 were injected with 1 ml BrdU (Invitrogen, Carlsbad, CA, USA) per 100 g body weight intraperitoneally. After 2 h, embryos were removed, trimmed and fixed with 4% paraformaldehyde. Antibody detection of BrdU involved standard immunostaining protocols. The proportion of BrdU-positive cells to total cells in the palatal shelf mesenchyme and epithelium were counted from five randomly selected sections per sample. Five palate samples of 3–5 dams were evaluated from each experimental group.
Apoptotic cells were assayed using TUNEL by use of the FragEL DNA Fragmentation Detection Kit, Fluorescein (Calbiochem, Darmstadt, Germany).
Primary cell culture and related assays
Primary mouse embryonic palatal mesenchymal (MEPM) cell culture was performed as described previously.12 Cells from passages 3–5 were used. For western blot analysis, protein samples from treated cells were boiled in SDS loading buffer and then subjected to SDS-PAGE following standard protocol.
For real-time quantitative PCR, total RNA was isolated from RA-treated MEPM cells using Trizol (Life Technologies, Carlsbad, CA, USA) and then reverse transcribed to cDNA using a PrimeScript RT reagent Kit (TaKaRa, Dalian, China). The PCR reaction system was prepared with a kit (SYBR Premix Ex Taq, TaKaRa), and the reaction was carried out on a real-time PCR machine (Mx3000P, Agilent Stratagene, Santa Clara, CA, USA). The primer sequences for RA receptor (RAR) and retinoid X receptor (RXR) were as described previously.13
For cell viability assay, a CCK-8 (Cell Counting Kit-8; Dojindo, Nanjing, China) kit was used. Briefly, cells were seeded in 96-well microtiter plates. After different treatments for indicated time, 10 μl CCK-8 solutions were added to each well, and the plates were incubated for additional 4 h at 37 °C. The absorbance was read at 450 nm on an automatic microwell plate reader (Bio-Rad, Berkeley, CA, USA).
Analysis of caspase-3 activities was performed using Caspase Apoptosis Detection Kit (Santa Cruz Biotechnology). Briefly, cells (1 × 106) were pelleted using centrifugation, washed with PBS two times and incubated in 500 μl lysis buffers on ice for 10 min, then 1 × reaction buffer and 10 μl DEVD-AFC substrate were added to lysis buffer. The reaction mixtures were incubated at 37 °C for 60 min. Activities were measured using spectrofluorometry. Enzymatic activities in cell lysate are directly proportional to the color reaction.
Statistical analysis
Data were analyzed by use of Student’s t-test or one-way ANOVA (SPSS 13.0). Results are considered significant at P<0.05. Each experiment was at least performed in triplicate.
Abbreviations
- E:
-
embryonic day
- MEE:
-
medial edge epithelium
- MEPM:
-
mouse embryonic palatal mesenchymal
- RA:
-
retinoic acid
- RAR:
-
retinoic acid receptor
- RXR:
-
retinoid X receptor
References
Ross SA, McCaffery PJ, Drager UC, De Luca LM . Retinoids in embryonal development. Physiol Rev 2000; 80: 1021–1054.
Wang W, Kirsch T . Retinoic acid stimulates annexin-mediated growth plate chondrocyte mineralization. J Cell Biol 2002; 157: 1061–1069.
Lai L, Bohnsack BL, Niederreither K, Hirschi KK . Retinoic acid regulates endothelial cell proliferation during vasculogenesis. Development 2003; 130: 6465–6474.
Kochhar DM, Johnson EM . Morphological and autoradiographic studies of cleft palate induced in rat embryos by maternal hypervitaminosis A. J Embryol Exp Morphol 1965; 14: 223–238.
Abbott BD, Harris MW, Birnbaum LS . Etiology of retinoic acid-induced cleft palate varies with the embryonic stage. Teratology 1989; 40: 533–553.
Takekoshi S . The mechanism of vitamin A induced teratogenesis. J Embryol Exp Morphol 1964; 12: 263–271.
Yasuhara R, Yuasa T, Williams JA, Byers SW, Shah S, Pacifici M et al. Wnt/β-catenin and retinoic acid receptor signaling pathways interact to regulate chondrocyte function and matrix turnover. J Biol Chem 2010; 285: 317–327.
Von Gise A, Zhou B, Honor LB, Ma Q, Petryk A, Pu WT . WT1 regulates epicardial epithelial to mesenchymal transition through β-catenin and retinoic acid signaling pathways. Dev Biol 2011; 356: 421–431.
Neto A, Mercader N, Gomez-Skarmeta JL . The Osr1 and Osr2 genes act in the pronephric anlage downstream of retinoic acid signaling and upstream of Wnt2b to maintain pectoral fin development. Development 2012; 39: 301–311.
Chiquet BT, Blanton SH, Burt A, Ma D, Stal S et al. Variation in WNT genes is associated with non-syndromic cleft lip with or without cleft palate. Hum Mol Gen 2008; 17: 2212–2218.
Niemann S, Zhao C, Pascu F, Stahl U, Aulepp U et al. Homozygous WNT3 mutation causes tetra-amelia in a large consanguineous family. Am J Hum Genet 2004; 74: 558–563.
Hu X, Chen ZG, Mao XY, Tang SJ . Effects of phenytoin and Echinacea purpurea extract on proliferation and apoptosis of mouse embryonic palatal mesenchymal cells. J Cell Biochem 2011; 112: 1311–1317.
Szabova L, Macejova D, Dvorcakova M, Mostbock S, Blazickova S, Zorad S et al. Expression of nuclear retinoic acid receptor in peripheral blood mononuclear cells (PBMC) of healthy subjects. Life Sci 2003; 72: 831–836.
Cuervo R, Valencia C, Chandraratna RA, Covarrubias L . Programmed cell death is required for palate shelf fusion and is regulated by retinoic acid. Dev Biol 2002; 245: 145–156.
Degitz SJ, Francis BM, Foley GL . Mesenchymal changes associated with retinoic acid induced cleft palate in CD-1 mice. J Craniofac Genet Dev Biol 1998; 18: 88–89.
Zhang Y, Mori T, Iseki K, Hagino S, Takaki H et al. Differential expression of decorin and biglycan genes during palatogenesis in normal and retinoic acid-treated mice. Dev Dyn 2003; 226: 618–626.
Alsayed Y, Uddin S, Mahmud N, Lekmine F, Kalvakolanu DV, Minucci S et al. Activation of Rac1 and the p38 mitogen-activated protein kinase pathway in response to all-trans-retinoic acid. J Biol Chem 2001; 276: 4012–4019.
Hormi-Carver K, Feagins LA, Spechler SJ, Souza RF . All trans-retinoic acid induces apoptosis via p38 and caspase pathways in metaplastic Barrett's cells. Am J Physiol Gastrointest Liver Physiol 2007; 292: G18–G27.
Kam RK, Deng Y, Chen Y, Zhao H . Retinoic acid synthesis and functions in early embryonic development. Cell Biosci 2012; 2: 11.
Hilliard SA, Yu L, Gu S, Zhang Z, Chen YP . Regional regulation of palatal growth and patterning along the anterior-posterior axis in mice. J Anat 2005; 207: 655–667.
Choi JW, Park HW, Kwon YJ, Park BY . Role of apoptosis in retinoic acid-induced cleft palate. J Craniofac Surg 2011; 22: 1567–1571.
Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, Chen Y . Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development 2002; 129: 4135–4146.
Rice R, Spencer-Dene B, Connor EC, Gritili-Linde A, McMahon AP et al. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J Clin Invest 2004; 113: 1692–1700.
Alappat SR, Zhang Z, Suzuki K, Zhang X, Liu H, Jiang Rs et al. The cellular and molecular etiology of the cleft secondary palate in Fgf10 mutant mice. Dev Biol 2005; 77: 102–113.
Iwatsuki K, Liu HX, Grunder A, Singer MA, Lane TF et al. Wnt signaling interacts with Shh to regulate taste papilla development. Proc Natl Acad Sci USA 2007; 104: 2253–2258.
Ahn Y, Sanderson BW, Klein OD, Krumlauf R . Inhibition of Wnt signaling by Wise (Sostdc1) and negative feedback from Shh controls tooth number and patterning. Development 2010; 137: 3221–3231.
Cho SW, Kwak S, Woolley TE, Lee MJ, Kim EJ, Baker RE et al. Interactions between Shh, Sostdc1 and Wnt signaling and a new feedback loop for spatial patterning of the teeth. Development 2011; 138: 1807–1816.
Jin YR, Han XH, Taketo MM, Yoon JK . Wnt9b-dependent FGF signaling is crucial for outgrowth of the nasal and maxillary processes during upper jaw and lip development. Development 2012; 139: 1821–1830.
Laplante M, Sabatini DM . mTOR signaling at a glance. J Cell Sci 2009; 122: 3589–3594.
Acknowledgements
We thank Dr Zhenguo Chen and Professors Shenqiu Luo, Xiaochun Bai, Hong Shen for their thoughtful discussion and advice. This work was supported by the National Natural Science Foundation of China (81071589 and 81171834) and by the Natural Science Foundation of Guangdong Province, China (S2011010003058).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Stephanou
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Hu, X., Gao, J., Liao, Y. et al. Retinoic acid alters the proliferation and survival of the epithelium and mesenchyme and suppresses Wnt/β-catenin signaling in developing cleft palate. Cell Death Dis 4, e898 (2013). https://doi.org/10.1038/cddis.2013.424
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.424
Keywords
This article is cited by
-
The mechanisms governing mouse embryonic palate mesenchymal cells’ proliferation associated with atRA-induced cleft palate in mice: insights from integrated transcriptomic and metabolomic analyses
Archives of Toxicology (2023)
-
Transcriptome analysis of Xenopus orofacial tissues deficient in retinoic acid receptor function
BMC Genomics (2018)
-
An E2F1/MiR-17-92 Negative Feedback Loop mediates proliferation of Mouse Palatal Mesenchymal Cells
Scientific Reports (2017)
-
Novel insights into a retinoic-acid-induced cleft palate based on Rac1 regulation of the fibronectin arrangement
Cell and Tissue Research (2016)
-
Implications of the Wnt5a/CaMKII Pathway in Retinoic Acid-Induced Myogenic Tongue Abnormalities of Developing Mice
Scientific Reports (2014)
