
Abstract
Rapidity and specificity are characteristic features of proteolysis mediated by the ubiquitin-proteasome system. Therefore, the UPS is ideally suited for the remodeling of the embryonic stem cell proteome during the transition from pluripotent to differentiated states and its inverse, the generation of inducible pluripotent stem cells. The Trim-NHL family member LIN41 is among the first E3 ubiquitin ligases to be linked to stem cell pluripotency and reprogramming. Initially discovered in C. elegans as a downstream target of the let-7 miRNA, LIN41 is now recognized as a critical regulator of stem cell fates as well as the timing of neurogenesis. Despite being indispensable for embryonic development and neural tube closure in mice, the underlying mechanisms for LIN41 function in these processes are poorly understood. To better understand the specific contributions of the E3 ligase activity for the stem cell functions of LIN41, we characterized global changes in ubiquitin or ubiquitin-like modifications using Lin41-inducible mouse embryonic stem cells. The tumor suppressor protein p53 was among the five most strongly affected proteins in cells undergoing neural differentiation in response to LIN41 induction. We show that LIN41 interacts with p53, controls its abundance by ubiquitination and antagonizes p53-dependent pro-apoptotic and pro-differentiation responses. In vivo, the lack of LIN41 is associated with upregulation of Grhl3 and widespread caspase-3 activation, two downstream effectors of p53 with essential roles in neural tube closure. As Lin41-deficient mice display neural tube closure defects, we conclude that LIN41 is critical for the regulation of p53 functions in cell fate specification and survival during early brain development.
Similar content being viewed by others

Main
Stem cell differentiation is driven by self-reinforcing regulatory loops working in parallel at the transcriptional as well as the post-transcriptional level. Less is known about post-translational mechanisms, including possible contributions by the ubiquitin-proteasome system. The ubiquitin-proteasome system is a major pathway for regulated protein degradation and is known to modulate the stability and activity of diverse factors that control cell fate decisions.1 The E3 ubiquitin ligase LIN41 was initially discovered as a critical downstream target of the let-7 miRNA.2 Lin41 activity was shown to be restricted to self-renewing stem cells and to oppose the pro-differentiation role of let-7.2, 3 Moreover, reestablishment of Lin41 expression was recently shown to be a prerequisite for the de-differentiation of somatic cells during the generation of induced pluripotent stem cells.4
The molecular functions of LIN41 in stem cells are just beginning to be elucidated. Also known as TRIM71, LIN41 is a founding member of the Trim-NHL protein family.2 Unlike C. elegans, mammalian LIN41 proteins have been shown to possess two activities: RNA-binding and translational repression mediated by the C-terminal NHL domain4, 5, 6, 7, 8 and E3 ubiquitin ligase activity mediated by the N-terminal Tripartite motif (Trim).7, 9, 10 The Trim domain consists of a RING-type zinc finger, two B-Box zinc fingers and a coiled-coil domain.11
The mutational phenotypes of many of the mammalian Trim-NHL genes cluster to the nervous system.11 Mice deficient in Lin41 display a 100% penetrant failure of neural tube closure, together with generalized growth retardation and embryonic lethality.10, 12, 13, 14 A role for LIN41 in neurogenesis is suggested by defects in gene expression during neural commitment by LIN41-deficient ES cells14 and the premature differentiation of neural progenitors in the CNS of LIN41-deficient embryos.10 The FGF-signaling intermediate SHCBP1 was identified as a substrate for LIN41-mediated ubiquitination in embryonic neurogenesis.10 To better characterize the E3 ubiquitin ligase activity of LIN41, we applied a proteomic approach to determine the effects of LIN41 on global ubiquitination patterns in pluripotent ES cells and in ES cells undergoing neural differentiation. Among a wealth of candidate substrates for LIN41-mediated ubiquitination, the tumor suppressor protein p53 was one of the most strongly affected proteins. Apart from its better-known role as ‘guardian of the genome’ in response to stress, a number of studies have described the sensitivity of the embryonic nervous system to perturbations in p53 regulation.15, 16, 17, 18 In this study, we present a new node for p53 regulation by LIN41 that is specifically active during differentiation of ES cells and also during embryonic neurogenesis in vivo. We show that LIN41 interacts with p53 and promotes p53 ubiquitination. As a consequence, multiple p53-dependent pro-apoptotic and pro-differentiation responses are reciprocally affected by either sustained LIN41 expression in differentiating ES cells or LIN41 deficiency in Lin41−/− ES cells or embryos. Taken together, our results indicate that LIN41 is required to dampen p53 responses and limit the extent of apoptosis during neural tube closure and early brain development.
Results
LIN41 and global ubiquitination in ES cells
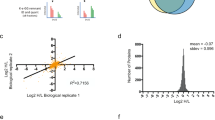
To identify direct substrates for LIN41, we analyzed global ubiquitination patterns in a previously described stable ES cell line (iLin41 cells) that allows doxycycline (Dox)-inducible expression of LIN41.5 We used this system to manipulate LIN41 expression in two distinct cell states: (1) pluripotent cells grown in the presence of leukemia inhibitory factor (LIF), and (2) cells undergoing neural differentiation for 3 days in response to retinoic acid (RA) (Figures 1a and b). Using the so-called UbiScan method to quantify ubiquitinated peptides (Figure 1a), we identified ~11 500 non-redundant diglycine-modified peptides within 3549 proteins (Supplementary file 1).

UbiScan for global identification of LIN41 ubiquitination substrates. (a) Proteomic strategy for global identification of LIN41-mediated ubiquitination targets under self-renewal conditions (LIF) and during neural differentiation with RA using a mouse ES cell line with Dox-inducible expression of LIN41-Flag. Each extract was processed by digestion with trypsin followed by affinity purification using an antibody specific for lysine residues carrying a diglycine linkage (K-GG). Enriched ubiquitin-modified peptides were analyzed by spectrometry. (b) iLin41 protein extracts prepared for the UbiScan were immunoblotted with α-LIN41 and α-OCT-4 antibodies to validate LIN41 induction by Dox and the conversion of pluripotent ES cells to differentiated cells after 3 days of RA treatment, respectively. (c) Venn diagram compares the sets of proteins showing a ≥2.5-fold increase in ubiquitination after LIN41 induction between the pluripotent and differentiated cell states. (d and e) Functional annotation analysis depicting significantly enriched Panther pathways among proteins with ≥2.5-fold more ubiquitination upon LIN41 induction in pluripotent (d) and differentiated (e) cells. Similar results were obtained for enrichment of p53-related KEGG or REACTOME pathways using Gene Set Enrichment Analysis (http://software.broadinstitute.org/gsea/msigdb/index.jsp, data not shown). (f) Diagram summarizing selected p53 pathways with LIN41 ubiquitination candidates from UbiScan depicted in blue and their functional relationships (arrow=direct activation, dotted arrow=indirect activation, inhibition line=direct negative regulation). (g) Fold change ratio of Dox+ versus Dox− in differentiated cells for the indicated diglycine-modified sites within p53 and schematic of p53 domain structure. DBD, DNA Binding Domain; NLS, Nuclear Localization Sequence; TAD, Transactivation Domain; TET, Tetramerization domain. See also Supplementary Figure 1 and Supplementary files 1–4
In pluripotent cells, 255 proteins showed a ≥2.5-fold increase in ubiquitination upon LIN41 induction (Supplementary file 2). Fifty-six of these proteins were also more ubiquitinated in response to LIN41 in differentiated cells among a total of 274 proteins (Figure 1c, Supplementary Figure 1 and Supplementary file 3). We performed functional enrichment analysis of these two sets of LIN41 responsive proteins to prioritize potential candidates for LIN41-mediated ubiquitination (Figures 1d and e). Of particular interest was the over-representation of p53-related pathways in differentiating cells (P<0.005 = 0.002; Figures 1e and f). In this study, we focus on regulatory interactions between LIN41 and p53 based on their opposing roles in regulating pluripotency and cellular reprogramming.
p53 is a potential substrate for LIN41-mediated ubiquitination
Among the LIN41 responsive proteins p53 was one of the five most strongly affected proteins in cells undergoing neural differentiation (Supplementary file 4). Multiple ubiquitination sites were detected within the p53 protein (Supplementary file 1). Induction of LIN41 in RA-treated cells resulted in a three- to tenfold increase in diglycine modification at lysines K313–315 (Figure 1g). These residues lie in a bipartite nuclear localization sequence positioned between the DNA binding and tetramerization domains (Figure 1g). Ubiquitination at position K364 and K299 was also increased more than twofold (Figure 1g). Together, these sites account for more than 90% of all recovered p53-derived peptides (Supplementary file 1).
Validating the effects on p53 by western blotting, we found that induction of LIN41 in RA-treated cells led to a significant reduction in p53 protein levels not seen in cells maintained in LIF (Figures 2a and b). This LIN41-dependent decrease in p53 was prevented by pretreatment with the proteasome inhibitor MG132. LIN41 induction also enhanced accumulation of higher molecular weight signals representing modified p53 in the presence of MG132 (Figures 2a and c). Consistent with the data from UbiScan, this impact of LIN41 on p53 was more prominent in differentiated cells. p53 mRNA levels, in contrast, did not change in response to LIN41 despite the differences seen in protein levels (Figure 2d).

LIN41 negatively regulates p53 protein levels by ubiquitination (a) Induction of LIN41 in differentiated, but not in pluripotent, ES cells downregulates endogenous p53 protein levels (compare lane 5 with 6 and lane 7 with 8) in a proteasome-dependent manner (compare lanes 3 and 4 with lanes 7 and 8). Expression levels of p53, LIN41, OCT-4 and GAPDH were determined by immunoblotting. A short and long exposure are shown for the p53 blot. (b and c) Quantification of unmodified (b) or ubiquitin-modified (c) p53 from LIN41-induced (Dox+) relative to untreated cell extracts. Relative band intensity was quantified from immunoblots, cell treatments as in a. Data from three independent experiments are presented as means±S.D. values. *P<0.05, **P<0.01, ***P<0.001. (d) LIN41 induction does not affect p53 transcript levels in either pluripotent or differentiated ES cells. p53 mRNA was quantified by qRT-PCR using Oaz1 mRNA for normalization. Data from three independent experiments are presented as means±S.D. values. (e) p53 is reduced in response to LIN41 induction during SFEBq cortical organoid differentiation. iLin41 organoid cultures were differentiated with and without Dox. p53 and LIN41 protein levels were monitored at indicated time points by immunoblotting. (f) LIN41 induction during neural differentiation promotes p53 poly-ubiquitination in vivo. Multi- and poly-ubiquitinated proteins from pluripotent and differentiated iLin41 cells were enriched using TUBEs after p53 induction with 1 μM doxorubicin and 4 h MG132 treatment. Ubiquitinated p53, auto-ubiquitinated LIN41 and pull-down efficiency were monitored by immunoblotting. (g) Mono- and poly-ubiquitinated proteins were purified using α-Ub-FK2-agarose beads capable of detecting all linkage types. iLin41 cells were prepared as for TUBE assay but without p53 induction. Western blots of input lysates and eluates were probed with a goat α-p53 antibody. Immunoblots of LIN41 and OCT-4 were included as positive and negative control, respectively. LIN41 induction increases ubiquitination levels of endogenous p53
We next studied the effects of sustained Lin41 expression in a more physiological neural differentiation paradigm that does not require chemical inducers such as RA: Serum Free Embryoid Body culture with quick aggregation (SFEBq).19 Similar to RA-induced differentiation, we observed a reduction in p53 levels in response to LIN41 overexpression throughout the early phase of neural differentiation (Figure 2e, full scan see Supplementary Figure 4e).
LIN41 promotes ubiquitination of p53 in response to RA
We used two approaches to characterize the LIN41-mediated modification of p53: capture of K48 and K63 poly-ubiquitinated proteins by tandem ubiquitin binding entities (TUBE), and immunoprecipitation (IP) with an α-ubiquitin antibody capable of detecting mono- as well as poly-ubiquitinated proteins irrespective of linkage type. For TUBE-mediated capture, we first treated all cultures with doxorubicin to induce p53. Under these conditions, LIN41 enhanced overall p53 ubiquitination (Figure 2f). The higher sensitivity of α-ubiquitin IP allowed us to confirm these results without doxorubicin treatment. As in the TUBE assay, LIN41 increased the intensity of ubiquitinated p53 bands (Figure 2g). Once again, the effect was markedly stronger in differentiated cells, as predicted by UbiScan. As a negative control, a poly-ubiquitination ladder detected after probing for OCT-4 was unaffected by exogenous LIN41.
Evidence for direct physical interaction between LIN41 and p53
LIN41 localizes in the cytoplasm in ES cells,5, 9 both nuclear and cytoplasmic localization have been reported for p53.20, 21, 22 Using the monoclonal anti-p53 Pab421 antibody, we were able to confirm partial overlap of the two proteins in the cytoplasmic compartment (Figure 3a). We next performed IP experiments in heterologous cells after co-transfection of epitope-tagged LIN41 and p53. Exogenous p53 was co-precipitated by Flag-tagged LIN41 and vice versa (Supplementary Figures 2a and b). To confirm this interaction, we next performed asymmetrical IPs in which an epitope-tagged protein was used to immunopurify an endogenous interaction partner. Flag-tagged LIN41 co-purified endogenous p53 and the positive control MOV10 in HEK293 cell extracts (Figure 3b). In reciprocal experiments, Flag-tagged p53 could interact with endogenous LIN41 in P19 cells (Figure 3c). Importantly, we could also demonstrate association between the endogenous proteins in ES cells based on co-IP of LIN41 using a highly specific antibody trap for p53 (Figure 3d).

LIN41 co-localizes and physically interacts with p53. (a) iLin41 cells were immunostained for endogenous p53, LIN41 and nuclear marker Draq5. (b) Flag-tagged LIN41 co-precipitates endogenous p53 in HEK293 cells. Co-precipitation of endogenous proteins after IP with either control IgG or α-Flag agarose was demonstrated by western blotting. (c) Flag-tagged p53 co-purifies endogenous LIN41. P19 cells were transfected with Flag-p53 or positive control Flag-Mov10. After α-Flag-IP, LIN41 co-purified with similar efficiency using either MOV10 or p53 as bait. (d) p53 from mouse ES cells was affinity purified using a mouse-specific p53 Trap. A human-specific p53 Trap served as negative control. Endogenous interaction was observed between p53 and LIN41, but not with GAPDH. (e) Schematic representation of LIN41 domains and the deletion constructs used to map the p53 interaction. (f) p53 interacts with the NHL domain of LIN41. LIN41-GFP fusion proteins containing the full-length protein or deletion constructs without the NHL, the RING motif or with only the NHL domain were co-expressed with Flag-tagged p53 in HeLa cells. GFP was included as a negative control. Input and bound proteins after Flag-IP were analyzed by western blotting with the indicated antibodies. (g) p53 directly interacts with LIN41. GST pull-down assays of in vitro synthesized LIN41 using GST only, p53-GST fusion protein or MDM2-GST. The presence of LIN41 in bound and unbound fractions was analyzed by immunoblotting. See also Supplementary Figure 2
We then performed a deletion analysis to define the domain in LIN41 responsible for p53 binding. Co-IP assays revealed that the NHL domain of LIN41 is necessary and sufficient for p53 binding (Figures 3e and f). To test for direct interaction, we analyzed whether LIN41 protein synthesized in vitro would interact with a p53-GST fusion protein. LIN41 co-purified with the p53-GST protein but not with either GST or MDM2-GST (Figure 3g). Taken together, these results demonstrate that p53 and LIN41 can interact in the absence of potential bridging factors from ES cells.
LIN41 affects proliferation during neural differentiation
Previous studies have shown that experimental reduction of p53 levels in neural stem cells leads to excessive proliferation, reduced apoptosis and impaired differentiation.23, 24 To investigate whether LIN41 negatively affects these p53 functions, we first examined the impact of LIN41 on cell proliferation. Induction of LIN41 increased cell numbers in RA-treated cells but not in undifferentiated cells (Figures 4a and b). Similar results were obtained by measuring incorporation of BrdU as an assay for proliferation (Supplementary Figure 3a). The effect of LIN41 on proliferation was associated with a small but significant shift toward the S and G2 phases after 3 days of RA-induced differentiation (Supplementary Figures 3b and c). Although this result is consistent with cell cycle changes reported in ES cells depleted for LIN41,5 the effect is likely too small to fully account for the difference in cell numbers we observed.

LIN41 affects proliferation and p53-dependent apoptosis during ES cell differentiation. (a and b) LIN41 differentially promotes proliferation in differentiating compared to pluripotent cell states. Dox-inducible iLin41 cells were cultured under proliferative (a) or differentiation conditions (b). Cell number was determined at 24 h intervals in triplicates. Data are presented as means±S.D. of three independent experiments. *P<0.05. (c) CASP3 activation during neural differentiation is p53 dependent. p53 WT and p53-deficient mES cells were cultured under proliferative or differentiation conditions. Cleaved CASP3 levels were determined by immunoblotting. (d) Induction of LIN41 during neural differentiation reduces CASP3 activation. Cleaved CASP3, p53, LIN41 and GAPDH levels of pluripotent and differentiated iLin41 cells without or with Dox treatment were compared by immunoblotting. (e) LIN41 inhibits p53-mediated apoptosis in response to stress. iLin41 cells differentiated in the presence and absence of Dox were pulse treated with 1 μM doxorubicin. p53 levels and CASP3 activation were determined by immunoblotting. (f) LIN41 induction in RA-differentiated stem cells reduces levels of p53 target mRNAs. Pluripotent and differentiated populations of iLin41 cells were cultured without or with Dox. mRNA levels of direct p53 transcriptional targets (Puma and Noxa) or the non-transcriptional target (Bim) within the apoptosis pathway were analyzed by qRT-PCR and normalized to Oaz1. Data are presented as mean values±S.D. from three independent experiments. *P<0.05, ***P<0.001. (g) LIN41 counteracts p53’s transactivation capacity in response to stress. iLin41 cells differentiated for 3 days in the absence or presence of Dox were pulse treated with 1 μM doxorubicin. mRNA levels of Puma, Noxa and Bim were measured by qRT-PCR assays and normalized to Oaz1. Data are presented as mean values±S.D. from at least three independent experiments. **P<0.01, ***P<0.001. See also Supplementary Figures 3 and 4
LIN41 induction inhibits apoptosis during ES cell differentiation
Visual inspection of cells over the course of differentiation suggested that the LIN41-dependent increase in cell numbers following RA treatment might be a consequence of greater cell viability and reduced cell death (Supplementary Figure 4a). Therefore, we hypothesized that the decrease in p53 levels associated with enforced LIN41 expression might protect differentiating cells from p53-dependent cell death pathways. Using activated caspase-3 (CASP3) as a marker for apoptosis, we found that induction of neural differentiation was associated with significant accumulation of cleaved CASP3 (Figures 4c and d). Using p53-deficient ES cells,22 we could show that activation of CASP3 in response to RA stimulation is p53 dependent (Figure 4c and Supplementary Figure 4b). Induction of LIN41 in RA-treated cells strongly inhibited CASP3 activation (Figure 4d and Supplementary Figure 4c), without affecting total CASP3 levels (Supplementary Figure 4f). Importantly, this effect was not limited to RA treatment and was also seen in two additional differentiation conditions: spontaneously differentiating cells 3 days after LIF withdrawal as well as SFEBq cultures (Supplementary Figures 4d and e). To directly test whether LIN41 can antagonize pro-apoptotic p53 function, we induced the DNA damage response in differentiating cells with the topoisomerase II inhibitor doxorubicin. As expected, p53 and activated CASP3 accumulated strongly in response to doxorubicin in wild type but not in p53-deficient ES cells, indicating that this apoptotic response is dependent on p53 (Supplementary Figures 4g and h). Importantly, LIN41 induction strongly reduced the p53 accumulation and CASP3 activation triggered by doxorubicin in iLIN41 cells (Figure 4e).
The ability of p53 to promote cell death is based in part on direct transcriptional activation of the pro-apoptotic factors Puma and Noxa. Steady-state levels of Puma and Noxa mRNA were significantly reduced upon LIN41 induction in pluripotent cells (Figure 4f). This decrease was stronger in differentiated cells, consistent with the stronger effect of LIN41 on p53 in these cells. The levels of Bim mRNA, a p53-independent pro-apoptotic gene, were not affected. We then asked if LIN41 can inhibit the transcriptional response to doxorubicin. Stress-dependent upregulation of Puma and Noxa mRNA levels was significantly attenuated in differentiated cells upon LIN41 induction (Figure 4g). Taken together, these results suggest that LIN41 interferes with p53-dependent pathways that promote cell death during ES cell differentiation.
Loss of LIN41 elevates p53 steady-state levels and reduces p53 ubiquitination
We next determined the effect of Lin41 deficiency on p53 in ES cells. For this purpose, multiple independent Lin41−/− and Lin41+/− ES cell lines were derived from knockout and heterozygote blastocysts from Lin41-deficient mice.13 In three independent experiments using three independent heterozygote and mutant clones each we observed an increase in steady-state p53 protein levels in the knockout ES cells (Figure 5a). Despite this increase in p53 protein, p53 mRNA levels were unchanged (Figure 5b). Downstream of p53 we detected significant upregulation of Noxa, but not Puma in Lin41 knockout ES cells (Figure 5c). The reason for this selective increase of downstream effectors upon loss of LIN41 is unclear, although there are several precedents for uncoupling of p53 levels and transcriptional activity in pluripotent cells.25

Loss of LIN41 elevates p53 steady-state levels and reduces p53 ubiquitination. (a) Comparison of p53 expression in undifferentiated wild type, heterozygote and homozygote Lin41-mutant ES cells shows a gene dose-dependent increase of p53 protein levels in Lin41-deficient cells. (b) Lin41 deficiency in mouse ES cells does not affect p53 transcript levels analyzed by qRT-PCR. Expression levels were normalized to Oaz1 mRNA. Data are presented as means±S.D. values from three independent clonal ES cell lines per genotype. (c) Constitutive levels of the p53 effector Noxa are increased in Lin41-deficient ES cells. Expression levels of Puma, Noxa and Bim were measured by qRT-PCR and normalized to Oaz1. Data are presented as means±S.D. values from three independent clonal ES cell lines per genotype. **P<0.01. (d) Cre-induced disruption of LIN41 in Lin41fl/fl; Rosa26-CreERT2 mES cells upregulates p53 steady-state levels. Conditional Lin41 knockout cell lines were cultured under proliferative and differentiation conditions. For the induction of Cre-mediated recombination, cells were treated with 500 nM 4-hydroxytamoxifen. p53, OCT-4 and LIN41 protein expression were examined in untreated cells (lanes 1 and 5), in two independent cell lines with stable Lin41 deletion (lanes 2 and 6 plus 3 and 7) or acute loss of Lin41 (lanes 4 and 8). (e) Quantification of p53 levels in two independent lines with stable Cre-induced Lin41 deletion relative to untreated cells. Relative band intensity was quantified from immunoblots from three independent experiments. Data are presented as means±S.D. values. *P<0.05, ***P<0.001. (f and g) Loss of LIN41 reduces p53 ubiquitination in vivo. After 4 h MG132 treatment, total ubiquitinated proteins from 2-day RA-treated Lin41fl/fl; Rosa26-CreERT2 cells were enriched using α-Ub-FK2-agarose beads or TUBEs, as indicated. Ubiquitination levels of endogenous p53, LIN41 and pull-down efficiency were detected by immunoblotting
To further confirm the results obtained with constitutive Lin41−/− ES cell lines, we tested p53 steady-state levels in a Lin41fl/fl; Rosa26-CreERT2 mES cell model, which allows Cre-mediated Lin41 disruption.14 Both acute and stable deletion of LIN41 lead to an increase in p53 protein levels (Figure 5d). Consistent with our overexpression system the LIN41-dependent effect on p53 is more pronounced in cells undergoing differentiation (Figure 5e). To determine if loss of LIN41 leads to a corresponding reduction in p53 ubiquitination, we performed TUBE and ubiquitin IP assays on Lin41fl/fl and Lin41−/− ES cell lysates. Using either method, LIN41 deletion in differentiating mES cells results in a reduction of p53 ubiquitination (Figures 5f and g). Altogether, our results with these two independent loss-of-function models further strengthen the hypothesis that LIN41 may be a new negative regulator of p53.
Widespread apoptosis in Lin41-deficient embryos
Deletion of LiN41 in the mouse leads to embryonic lethality and failure of cranial neural tube closure10, 12, 13, 14 (Figure 6a). Likewise, successful neural tube closure is sensitive to defects in p53 expression and regulation.15, 16, 17, 26, 27 As either loss of Lin41 or dysregulation of p53 causes NTD, we asked whether p53 function might be affected in Lin41 knockout embryos. Immunostaining for p53 revealed increased nuclear levels of p53 in the neuroepithelium of mutant embryos compared to heterozygote littermates at E10.5 (Figures 6b and c).

Increased levels of p53 during brain development in the absence of Lin41. (a) Lin41 is essential for cranial neural tube closure. Failure of neural tube closure (asterisk) and growth retardation in Lin41−/− embryos in comparison to control Lin41+/+ littermate at E9.5 gestation. (b) Upregulated p53 expression during neural tube closure of Lin41-deficient embryos. Transverse cranial sections of E10.5 heterozygote and homozygote knockout Lin41GT;lacZ transgenic embryos were processed for immunohistochemistry, results for p53 and DAPI are shown. Lin41 mutants display neural tube closure defects (asterisk). (c) Higher magnification of b to better visualize p53 and LIN41 expression in the neuroepithelium. Lin41-deficient embryos exhibit increased nuclear levels of p53. In merged views, p53 is shown in green, β-galactosidase (β-Gal) from the Lin41-gene trap allele in red and DAPI in blue
Considering that LIN41 induction strongly inhibited p53-dependent apoptosis during ES cell differentiation, we assayed the levels of activated CASP3 in vivo. In contrast to heterozygote embryos, Lin41−/− littermates exhibited extensive cell death in the neuroepithelium and surrounding mesenchyme (Figure 7a, Supplementary Figures 5a and b). The increase in cell death within the Lin41-expressing neuroepithelium was confirmed at higher magnification (Figure 7bii, iii) and was particularly prominent in the vicinity of the optic vesicles (Figures 7bi). These results show that loss of LIN41 results in aberrant p53 activation and massive programmed cell death during the period of neural tube closure.

LIN41 affects embryonic apoptosis and the expression of the pro-differentiation gene Grhl3. (a) Widespread apoptosis during neural tube closure of Lin41-deficient embryos. Transverse cranial sections of E10.5 heterozygote and homozygote knockout Lin41GT;lacZ transgenic embryos were processed for immunohistochemistry, results for activated CASP3 and DAPI are shown. Lin41 mutants display accumulation of CASP3-positive cell clusters. (b) Higher magnification of a to better visualize apoptosis in the optic vesicle (i) and neuroepithelium (ii–iii) of Lin41-deficient embryos. In merged views, activated CASP3 is shown in red, β-galactosidase (β-Gal) from the Lin41-gene trap allele in green and DAPI in blue. (c) Grhl3, but not Grhl2, is responsive to p53 induction in ES cells undergoing neural differentiation. qRT-PCR analysis of Grhl2 and Grhl3 mRNAs following doxorubicin treatment. Lin41 and p53 transcript levels are unaffected, as expected. (d) LIN41 induction in RA-differentiated stem cells reduces levels of Grhl3 mRNAs. Pluripotent and differentiated populations of iLin41 cells were cultured without or with Dox. mRNA levels of Grhl3 were analyzed by qRT-PCR and normalized to Oaz1. (e) LIN41 loss of function in mouse E9.5 embryos leads to elevated Grhl3 mRNA levels. Grhl3, Lin41 and p53 transcript levels were analyzed by qRT-PCR and normalized to Oaz1. All data are presented as mean values±S.D. from three independent experiments. *P<0.05, **P<0.01, ***P<0.001. See also Supplementary Figure 5
LIN41 modulates Grainyhead-like 3 expression
The transcription factor Grainyhead-like 3 (GRHL3) is a p53 target gene28 that is associated with NTDs and cell fate specification.29, 30, 31 To determine if LIN41 affects p53 responses apart from the classical anti-apoptotic targets Puma and Noxa, we tested Grhl3. We first confirmed that Grhl3, but not the related Grhl2, is a strong p53 transcriptional target in our ES cell system by treating iLin41 cells with RA for 3 days followed by induction of p53 with doxorubicin (Figure 7c). Grhl3 levels were significantly reduced in response to LIN41 induction specifically in differentiated cells (Figure 7d). Conversely, Grhl3 transcript levels were elevated in Lin41−/− embryos compared to Lin41+/+littermates, while Lin41+/−heterozygotes displayed intermediate levels (Figure 7e). In both the Drosophila and mouse nervous systems, Grainyhead family transcription factors are responsible for the specification and terminal differentiation of specific stem cell populations.30, 32 Therefore, by regulating the extent of apoptosis and the cell fate determinant Grhl3, LIN41 appears to act via two distinct but potentially complementary processes. A schematic model summarizing our results for LIN41 activity vis-à-vis p53 is presented in Figure 8.

Working model for the interplay of LIN41 and p53 during neural commitment and neural tube closure. Physical interaction between LIN41 and p53 leads to ubiquitination and inactivation of p53 in cells undergoing neural differentiation. By targeting p53, LIN41 antagonizes pro-apoptotic (CASP3) and pro-differentiation effectors (Grhl3), two pathways downstream of p53 with essential roles in neural tube closure. These results indicate that LIN41 is vital to dampen p53 responses and limit the extent of apoptosis in both ES cell differentiation and mouse embryogenesis, which may account for the embryonic lethality and NTDs of Lin41-Null mice
Discussion
LIN41 is a member of an emerging class of RNA-binding proteins with E3 ubiquitin ligase activity. We report the results of an unbiased, global screen for potential targets of the E3 ubiquitin ligase activity of LIN41 that led us to investigate the role of LIN41 in the regulation of p53 in ES cells. Several lines of evidence point to a connection between Trim-NHL E3 ligases and p53. The first was the finding that TRIM3 is a direct transcriptional target of p5333 that in turn mediates degradative ubiquitination of the p53 response gene product CDKN1a/p21.34 Like TRIM3, TRIM32 is a transcriptional target of p53 but feeds back directly by targeting p53 itself for degradative ubiquitination.35 The first link between LIN41 and the p53 pathway was the recent finding that LIN41 mediates miRNA-dependent post-transcriptional silencing of the Cdkn1a mRNA, thereby limiting checkpoint control of the cell cycle in ES cells.5
Given these precedents, we were struck by the enhancement of p53-derived peptides in RA-treated cells seen in the UbiScan. The most strongly affected sites (K313–K315) are within the nuclear localization sequence of p53, a prominent site for the regulation of p53 activity in ES cells.21, 36 Furthermore, a knock-in mouse expressing p53 mutated for these three residues exhibits highly penetrant developmental abnormalities including exencephaly.37 It has proven difficult to assign specific functions to individual lysines in p53,36 however, ubiquitination of these lysines by MDM2 has been shown to interfere with nuclear import.38 For LIN41, our results suggest a function in degradative poly-ubiquitination, as indicated by the reduction in p53 protein levels in response to ectopic LIN41 and their increase in Lin41-deficient ES cells. However, the pattern of modified bands we see in ubiquitin pull-down assays is also consistent with mono- and oligo-ubiquitination. At present, we can also not exclude the possibility that LIN41 does not act alone or directly to enhance p53 ubiquitination. In addition to potential cooperation with other E3 and E4 ubiquitin ligases, LIN41 might also affect other post-translational modifications such as acetylation of K31439 or phosphorylation of p53 by the stem cell-specific activity of Aurora kinase A at S312.40 Alternatively, Aurora kinase A-mediated phosphorylation adjacent to the major ubiquitinated lysines in ES cells (K313–K315) might interfere with p53 ubiquitination. Thus, loss of Aurora kinase A activity after LIF withdrawal40, 41 might be responsible for the enhanced activity of LIN41 toward p53 in differentiating ES cells. Further work will be required to more precisely define how LIN41 fits into the network of p53 post-translational modifications.
p53 is best known as a crucial tumor suppressor that marshals the cellular stress response. Nevertheless, evidence from many studies indicates that p53 regulates cell cycle exit, metabolism and various other aspects of neural development and differentiation in the absence of acute stress.42 Of particular relevance, basal p53 activity was found to limit stem cell self-renewal, as p53-deficient neural stem cells display excess proliferation and reduced apoptosis.23, 24 In our experiments, LIN41 induction during neural differentiation led to a modest increase in the proliferation rate and cell cycle parameters and to a dramatic decrease in apoptosis, mimicking the properties of p53-deficient neural stem cells. We conclude that LIN41 acts to limit p53 activity, in particular p53-mediated apoptosis pathways, during neural differentiation of ES cells to control the balance between self-renewal, differentiation and cell death.
Approximately 20% of female p53-deficient embryos exhibit NTDs.17, 18 More severe developmental disturbances result from p53 hyperactivity due to loss or inactivation of negative regulators of p53. For example, a 100% penetrant failure of neural tube closure is observed after pharmacological stabilization of p5315 or mutational inactivation of the MDM2-associated co-factor MDMX.26 Thus, either loss of LIN41 or dysregulation of p53 cause NTDs, suggesting that LIN41 might be essential for the regulation of p53 functions during early brain development.
While there is some controversy regarding the importance of apoptosis for the normal progression of neural tube closure, excessive cell death is associated with failure of closure.31, 43, 44 We found that enforced LIN41 expression inhibited p53-dependent apoptosis during ES cell differentiation and, inversely, lack of LIN41 in E10.5 Lin41−/− embryos led to extensive cell death compared to wild type or heterozygote littermates. Previously, Chen et al.10 reported no significant increase in TUNEL staining of Lin41 mutant neural tubes at E9.5 or E10.5, although they noted increased apoptosis in the mutant hindbrain by E11.5. The activated CASP3 antibody we used, in contrast to the TUNEL assay, can detect cells in early phases of apoptosis and has been shown to be approximately threefold more sensitive than the TUNEL assay.45 Although strain-specific differences may also play a role, we were also able to detect a differential degree of apoptosis at E9.5, suggesting that CASP3 may be the more sensitive assay.
Besides antagonizing p53-dependent apoptotic pathways, LIN41 also modulates the expression of an additional p53 downstream effector, Grhl3. In the mammalian nervous system, Grhl3 coordinates fate specification of ectodermal progenitor cells during neurulation, a process essential for neural tube closure.30 As a consequence, Grhl3-deficient mice consistently display spinal closure defects associated with a low incidence of exencephaly.29, 46 Based on these convergent functions, we suggest that misregulation of p53 and the p53 effectors we have identified account for the growth deficit, embryonic lethality and NTDs of Lin41-deficient mice.
The reciprocal regulatory relationship between LIN41 and let-7 is increasingly recognized as an important genetic switch governing stem cell properties.2, 4, 9, 14, 47 However, considerably more is known about downstream effectors for let-7 than for LIN41. Despite progress in identifying substrates for the RNA-binding activity of LIN414, 5, 7, 8 there is a need for a better understanding of the conserved E3 ligase activity of LIN41 and the other Trim-NHL family members. In this study, we address this need and describe a new layer of post-translational stem cell regulation by LIN41: the ubiquitination and inhibition of the pro-differentiation and pro-apoptotic activities of p53. We suggest that one vital role of LIN41 is to restrain p53 activity to favor cell differentiation over cell death.
Materials and Methods
Plasmids
GFP-hAgo2 #11590, pFLAG/HA-Mov10 #10976, pcDNA3 flag p53 #10838, GFP-p53 #12091 and pFRT/FLAG/HA-DEST Eif2c2 #19888 used for transfection were purchased from Addgene Inc., Cambridge, MA, USA). peGFP-C1 (Clontech) and p3xFLAG-CMV-10 were transfected as negative controls for co-IP assays. Generation of p3xFLAG-Lin41 and Lin41-eGFP deletion mutant series were described in Rybak et al.9 For cloning of the T7-Lin41 construct, full-length Lin41 was amplified from the p3xFLAG-Lin41 vector using the NdeI-p3xFlag-CMV-10 F and C-CMV-24 Sequencing primer (for primer sequences, see Supplementary Table 1). The PCR fragment was digested by NdeI and BamHI enzymes and inserted in the DHFR control plasmid after removing the DHFR gene.
Mammalian cell culture and transfection
HEK293T (Human Embryonic Kidney) and HeLa (Human Cervix Carcinoma) cells purchased from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany) were grown as adherent monolayers in DMEM supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, 50 U/ml penicillin and 50 μg/ml streptomycin at 37 °C in a humidified incubator with 5% CO2. Plasmid transfection of HEK293T and HeLa cells was performed with calcium phosphate. Cells were seeded in a 10 cm culture dish 1 day before transfection to attain 90% confluence. Medium was replaced 2–4 h prior to transfection. A measure of 500 μl calcium–DNA solution, containing 30 μg plasmid DNA, 62.5 μl 2 M CaCl2 and MilliQ water, was added dropwise to an equal volume of 2 × HBS phosphate solution (280 mM NaCl, 1.5 mM Na2HPO4, 50 mM HEPES pH 7.13) with continuous vortexing. After a 30-min incubation at room temperature the precipitate was distributed dropwise over the cells under gentle agitation of the culture plate. Cells were incubated overnight under standard growth conditions before collecting for IP assays.
P19 embryonic carcinoma cells were maintained in α-MEM with 10% FBS, 50 U/ml penicillin and 50 μg/ml streptomycin. Cells were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Six hours prior to transfection 8 × 105 cells were seeded per well of a six-well plate in 2 ml Opti-MEM (Invitrogen) and 3% FBS without antibiotics. For each transfection 5 μl Lipofectamine was diluted in 150 μl of Opti-MEM without serum. In a second reaction tube, 2 μg DNA was diluted in the same volume of Opti-MEM. Subsequently, DNA and Lipofectamine solutions were mixed and kept for 20 min at room temperature allowing formation of liposome complexes before adding dropwise to the cells. After 16 h incubation under standard growth conditions, the DNA–lipid complexes were removed by changing to P19 complete growth medium. Cells were collected for further analysis 48 h post transfection.
ES cell culture and differentiation
Dox-inducible Flag-Trim71 KH2 ESC lines (iLin41) were a gift from Dr Richard Gregory.5 Induction of LIN41 expression was obtained by addition of 0.5 μg/ml Dox. This allows binding of the tetracycline-controlled transactivator rtTA to the tet operator and thus induces LIN41-Flag expression. The iLin41 mouse embryonic stem cell line was cultured under feeder free conditions on 0.2% gelatin-coated plates in DMEM (Gibco #21969-035, Carlsbad, CA, USA), supplemented with 20% FBS (PANSera, 2602-P272405, Pan-Biotech, Aidenbach, Germany), 1% L-glutamine, 20 mM HEPES (Gibco #15630114), 1 × MEM NEAA (Gibco #11140), 1% Pen/Strep, 0.1 mM β-mercaptoethanol (Gibco #31350) and 1000 U/ml LIF (#GSR-7001 MTI-GlobalStem, Gaithersburg, MD, USA) (ES cell medium). For general maintenance, iLin41 cells were grown to 70–80% confluency and passaged every 2 days at a ratio of 1 : 8.
To perform experiments with iLin41 cells under proliferative conditions, 7 × 105 cells were seeded on a 6 cm culture dish and cultured for 48 h in ES cell media containing LIF. For spontaneous differentiation by LIF removal, 105 cells were seeded on a 6 cm dish in ES cell medium. Eighteen hours later cells were washed once in ES cell medium without LIF and cultured for 72 h in ES cell medium without LIF. For neural differentiation using RA, 18 h after seeding (1.5 × 105 cells/6 cm) in ES cell medium, medium was replaced with ES cell medium without LIF supplemented with 5 μM RA and was replaced every 2 days. Cells reached 90% confluence after 3 days of RA treatment and were collected for further analysis.
The protocol for SFEBq19, 48 was used to induce cortical differentiation in iLin41 ES cells in the absence or presence of 2 μg/ml Dox. On Day 0, iLin41 cells were dissociated to a single-cell suspension in 0.25% (wt/vol) trypsin-EDTA and seeded (3000 cells in 100 μl/well) in cortical differentiation medium (GMEM from Gibco #11710-035, 1% MEM NEAA, 1% pen/strep, 1 mM sodium pyruvate, 10% knockout serum replacement from Invitrogen #10828028, 0.1 mM β-mercaptoethanol) in 96-well Lipidure-Coat U-bottom low-cell-adhesion plates (Amsbio #AMS.51011610, Abingdon, UK). After 2 days of differentiation and on consecutive days thereafter to Day 7, the resulting spheroid cell aggregates were collected for immunoblot analysis. For this purpose, eight aggregates per condition were transferred to PBS. After removing PBS, settled aggregates were lysed in 100 μl lysis buffer (100 mM Tris pH 6.8, 1% SDS, 25 mM EDTA, 25 mM EGTA, 1 × protease inhibitor cocktail) and sonicated using a Bioruptor for 10 cycles of 30 s on/30 s off at high intensity. A measure of 33 μl of 4 × SDS loading buffer was added directly to the lysate and heated at 95 °C for 5 min. An amount of 10 μg protein of the crude lysate was resolved by SDS-PAGE, followed by western blotting.
Multiple-independent Lin41−/− and Lin41+/− ES cell lines from knockout and heterozygote blastocysts from a mouse strain in which Lin41 is inactivated by a gene-trap insertion in the second intron13 were generated using standard procedures. Details are available upon request. These stem cell lines were generally maintained on a layer of feeder cells on 0.2% gelatin-coated plates in Advanced DMEM/F12 (Invitrogen #12634010) supplemented with 20% knockout serum replacer, 2 mM L-glutamine, 1% pen/strep, 0.1 mM β-mercaptoethanol and 1000 U/ml LIF. One day before seeding, 6 cm dishes were coated with 1 ml sterile-filtered 0.2% gelatin solution in PBS for at least 30 min and incubated at 37 °C. Mitomycin-C-treated CF1 MEF feeder cells (CellSystems ASF-1223) were seeded at a density of 2 × 105 cells per 6 cm dish in DMEM supplemented with 10% FBS, 2 mM glutamine, 1% pen/strep and grown overnight. The next day, 3.5 × 105 cells were seeded on the prepared MEF dishes, and medium was changed to ES cell medium with LIF. ES cells were split every second day for maintenance or collected 48 h after seeding.
The conditional Lin41 knockout mES cell line (Lin41fl/fl; Rosa26-CreERT2)14 was cultured under feeder free conditions on gelatin-coated plates in Knockout DMEM (Gibco #10829018) supplemented with 15% FCS, 1% MEM NEAA, 1 × Glutamax, 1% pen/strep, 0.1 mM β-mercaptoethanol and LIF. For stable Lin41 deletion, the Cre recombinase was induced using 500 nM 4-hydroxytamoxifen (Sigma, St. Louis, MO, USA) for 48 h. Cells were further cultured for at least five passages before use in experiments. Differentiation experiments were performed as described for iLin41 cells.
p53-deficient and parental mES cell lines were a gift from Dr Hitoshi Niwa.22 These cells were cultured without feeder cells on gelatin-coated dishes in Glasgow minimal essential medium (Gibco # 11710035) supplemented with 10% FBS, 1 mM sodium pyruvate, 0.1 mM β-mercaptoethanol, 1 × MEM NEA, 1% pen/strep and LIF.
Ubiscan and bioinformatic analysis
For the global screen of LIN41’s ubiquitination targets, a set of four extracts was prepared from Dox-inducible Flag-Trim71 KH2 mouse ES cells (iLin41)5 cultured in the presence or absence of Dox and maintained either 48 h in the pluripotent state in the presence of LIF or subjected to RA-induced differentiation for 3 days. Prior to cell lysis, cells were treated with 20 μM MG132 for 4 h. After two wash steps with 10 ml ice-cold PBS, cells were lysed by scraping in 10 ml urea lysis buffer (20 mM HEPES pH 8, 9 M Urea, 1 mM sodium orthovanadate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate). These lysates were further processed and analyzed for global ubiquitination profiling by Cell Signaling (UbiScan, Cell Signaling). For this purpose, equal amounts of protein extract were reduced with 4.5 mM DTT and alkylated with 10% iodoacetamide. After tryptic digestion with 10 μg/ml trypsin-TPCK (Promega, Madison, WI, USA) peptides were separated from non-peptide material by C18 solid-phase extraction prior to lyophilization. Peptides were affinity purified using an agarose-conjugated antibody specific for lysine residues carrying a diglycine linkage (K-GG) (Cell Signaling #8925). To desalt and concentrate the eluate, peptides were loaded onto a 10 cm × 75 μm PicoFrit capillary column packed with Magic C18 AQ reversed-phase resin. The column was developed with a 120-min linear gradient of acetonitrile in 0.125% formic acid delivered at 280 nl/min. Tandem mass spectra were collected with an LC-MS/MS analysis using LTQ-Orbitrap-Elite, ESI-CID (MS parameter settings: MS run time 120 min, MS1 scan range (300.0–1500.00), top 20 MS/MS (min signal 500, isolation width 2.0, normalized coll. energy 35.0, activation-Q 0.250, activation time 20.0, lock mass 371.101237, charge state rejection enabled, charge state 1+ rejected, dynamic exclusion enabled, repeat count 1, repeat duration 35.0, exclusion list size 500, exclusion duration 40.0, exclusion mass width relative to mass, exclusion mass width 10 ppm). MS/MS spectra were evaluated using SEQUEST 3G and the SORCERER 2 platform from Sage-N Research (v4.0, Milpitas CA).49 Searches were performed against the NCBI Mus musculus database, release date 28 June 2011, with mass accuracy of +/−50 ppm for precursor ions and 1 Da for product ions. Results were filtered with mass accuracy of +/–5 ppm on precursor ions and presence of the intended motif. The default false positive rate was set at 5%. The full results of the four samples analyzed in duplicates are provided in Supplementary file 1. The functional annotation tool of the database for annotation, visualization and integrated discovery v6.750 was used to identify significantly enriched Panther pathways (P≤0.05) among peptides with more than 2.5-fold ubiquitination.51 To prioritize potential candidates with highest ubiquitin modification upon LIN41 induction (Supplementary file 4), we increase confidence by filtering the PTMScan data based on fold change ≥2.5, max intensity >200 000 and CV <50%.
Western blots
Protein lysates were generally prepared from cells grown as monolayers. For urea lysates, cells were directly scraped in urea lysis buffer at room temperature. After 15 min incubation on ice, protein lysates were clarified by centrifugation at 12 000 × g, 4 °C for 15 min. Total protein concentration of crude lysates was routinely determined using Bradford assay (Bio-Rad Protein Assay, Hercules, CA, USA) with bovine albumin serum as calibration standard (0-1 μg/μl) at 595 nm wavelength in the photometer (Nanodrop). The separation of proteins was achieved in 6–15% polyacrylamide gels using the Bio-Rad SDS gel electrophoresis system. Subsequently, proteins were transferred to an ImmobilonP membrane (Millipore, Billerica, MA, USA) and incubated in blocking buffer (5% non-fat dry milk or 3% BSA in 1 × PBS-T). Dilutions of primary antibodies used are provided in Supplementary Table 2, incubation time was adjusted as required. After washing, horseradish peroxidase-conjugated secondary antibodies were applied for 1 h. Chemiluminescent detection was performed using ECL substrate Bio-Rad Clarity and the imaging system ImageQuant LAS (GE Healthcare, Little Chalfont, UK).
Ubiquitination assay
For in vivo ubiquitination assays, 4.5 × 106 iLin41 cells were grown on a 25 cm culture dish for 48 h in ES cell medium containing LIF as pluripotent stem cells. For differentiated cells, iLin41 cells were seeded 1 day before neural differentiation (1.2 × 106 per 25 cm plate) in ES cell medium and cultured 72 h in RA-containing medium. Prior to cell lysis, cells were pulse treated for 1 h with 1 μM doxorubicin to increase p53 protein levels. Subsequently, fresh medium with 20 μM proteasome inhibitor MG132 was added for 4 h to stabilize ubiquitinated proteins. Cells were washed with 10 ml PBS and collected directly by scraping in 1.5 ml TUBE lysis buffer (50 mM Tris-HCl pH 7.5, 0.15 M NaCl, 1 mM EDTA pH 8, 1% NP-40, 10% glycerol, 50 mM chloroacetamide, 50 mM NaF, 5 mM Na4P2O7, 10 mM β-glyceropyrophosphate, 20 μM MG132, 1 × protease inhibitor cocktail). Lysates were incubated 10 min on ice and clarified by centrifugation for 15 min at 14 000 × g. A measure of 30 μl of the homogenate was taken for the examination of the input by western blot. Subsequently, the lysates were precleared by incubation with 30 μl of 50% protein A/G beads slurry (Thermo Scientific, Waltham, MA, USA) for 30 min at 4 °C in a tube rotator. Precleared cell lysates were tumbled end-over-end overnight at 4 °C with 30 μl equilibrated TUBE-agarose 2 (Life Sensors, Malvern, PA, USA) beads. These affinity ubiquitin traps bind preferentially to proteins modified by K63 and K48 poly-ubiquitinated linkages. After IP, the resin was collected by low-speed centrifugation (1500 × g, 4 °C) for 5 min. Bound complexes were washed four times with TBS-T buffer and eluted with one bead volume of 2 × SDS reducing sample buffer by boiling for 5 min at 98 °C. The samples were centrifuged for 1 min at 13 000 × g at 4 °C. Supernatants were resolved by 10% SDS-PAGE, followed by immunoblotting with α-ubiquitin and substrate-specific antibodies.
As a second test for in vivo ubiquitination, we used a highly efficient α-multi-ubiquitin monoclonal antibody (clone FK2, MBL) covalently coupled to agarose beads. This antibody detects all linkage types of mono- and poly-ubiquitinated proteins. iLin41 cells were prepared as for the TUBE assay but without p53 induction. An amount of 2.5 mg total protein was added to 25 μl FK2 ubiquitin resin and tumbled overnight at 4 °C. After washing with TUBE buffer (all centrifugation steps were performed at 2500 × g for 10 s at 4 °C), bound proteins were eluted and analyzed by immunoblotting.
Ubiquitin assays with 2 days RA-stimulated Lin41fl/fl; Rosa26-CreERT2 cells were performed as described for iLin41 cells but without p53 induction.
Immunoprecipitation
α-Flag IPs to determine protein–protein interaction partners were performed from extracts of HEK293T, HeLa or P19 cells transiently expressing Flag-fusion constructs (p3XFLAG-Lin41, Flag-p53, pFLAG/HA-Mov10, pFRT/FLAG/HA-DEST Eif2c2, p3XFLAG-CMV-10). Cleared cell lysates of 90–95% confluent 10 cm diameter tissue culture plates were prepared in 200 μl non-denaturating co-IP buffer (2% Triton X, 1 × protease inhibitor cocktail in 1 × PBS, pH 7.4). A measure of 20 μl of the homogenate was taken for examination of the input by western blot. IP was carried out with the rest of the lysate. For that purpose, the volume of the protein suspension was brought to at least 500 μl with Co-IP buffer. Subsequently, the lysate was precleared by incubation with 30 μl of 50% protein A/G beads slurry (Thermo Scientific) for 1 h at 4 °C in a tube rotator. Precleared cell lysates were tumbled end-over-end overnight at 4 °C with 30 μl of α-Flag M2 agarose (Sigma). The immune complex was collected by centrifugation for 10 s at 10 000 × g, 4 °C and washed at least five times with ice-cold Co-IP buffer. Next, the beads were washed once more with 1 ml ice-cold PBS and the mixture was transferred in a new 1.5 ml tube before centrifugation. After removing the supernatant completely, 30 μl of 2 × SDS-PAGE loading buffer was added to the 15 μl of settled beads containing bound antigen. For elution, samples were then heated for 5 min at 95 °C, spun down and 25 μl/lane analyzed by western blotting.
Endogenous precipitation
For each endogenous IP 1.8 × 106 uninduced iLin41 mouse embryonic stem cells were cultured for 48 h in ES cell medium containing LIF in a 10 cm dish. After washing with PBS, cells were collected by scraping in 1 ml PBS and recovered by centrifugation at 6000 × g for 5 min at 4 °C. The cell pellet was resuspended in 200 μl ice-cold lysis buffer (1% Triton X-100, 1 × protease inhibitor cocktail in 1 × PBS) by passing five times through a 25-G syringe. After a 30-min incubation on ice, the lysate was cleared for 15 min at 13 000 × g at 4 °C. Ten percent of the homogenate was taken for the examination of the input. For the IP, the rest of the lysate was diluted with 300 μl STE buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA pH 8, 1 × protease inhibitor cocktail). Subsequently, 25 μl of equilibrated p53 Trap_A (Chromotek, Martinsried, Germany) bead slurry was added per reaction and incubated for 1 h at 4 °C. p53 C-term-Trap_A reacts with both human and mouse p53, whereas p53 N-term Trap_A only detects human p53 and therefore served as a control for unspecific binding. Beads were recovered by centrifugation at 2500 × g for 2 min at 4 °C, washed three times with STE buffer and resuspended in 30 μl 2 × SDS sample buffer for elution by boiling.
In vitro Protein synthesis
LIN41 protein for the in vitro interaction assays was generated using the PURExpress In Vitro Protein Synthesis Kit (New England Biolabs, Ipswich, MA, USA). For creating a Lin41 template containing the T7 promoter, ribosome-binding site and T7 terminator required for a one-step transcription and translation, the DHFR gene from the positive control plasmid of the kit was replaced with Flag-tagged full-length Lin41 cDNA. To remove residual RNaseA activity from plasmid DNA preparation, ethanol precipitation was carried out. An amount of 500 ng of the purified DNA in 10 mM Tris-HCl pH 8 was added to the in vitro expression reaction containing 10 μl solution A, 7.5 μl solution B, 20 units RNase inhibitor, ad 25 μl with nuclease-free H2O. After 3 h at 37 °C 25 μl glycerol was added to the reaction. A measure of 5 μl of the sample was loaded on a SDS gel to estimate the protein concentration by Coomassie staining with PAGEblue.
GST pull-down
For examination of direct protein interactions in vitro, GST pull-down assays were performed. A measure of 50 μl of glutathione-sepharose 4B beads (GE Healthcare) were equilibrated with GST buffer (50 mM NaCl, 50 mM Tris-HCl pH 7.5, 5 mM EDTA, 1% Triton X-100 and 1 × protease inhibitor cocktail). An amount of 1 μg GST (Sigma #SRP5348), 6 μg p53-GST (Enzo #BML-FW9370-0050) or MDM2-GST (Enzo #BML-UW0200-0025) was added to the beads in 800 μl GST buffer and incubated on a rotator for 1 h at 4 °C. The resin was then collected by low-speed centrifugation (500 × g for 20 s at 4 °C), washed once with 500 μl of complete GST buffer and twice with GST buffer without protease inhibitor cocktail. A measure of 10 μl of in vitro synthesized LIN41 protein were added to the GST-coupled matrix and tumbled end-to-end in 800 μl GST buffer for 2.5 h. After centrifugation, 30 μl of the supernatant was saved as unbound fraction for western blot analysis. The immunoprecipitates were washed three times with GST buffer without protease inhibitor cocktail, resuspended in one bead volume of 2 × SDS sample buffer and heated at 95 °C to release bound proteins for subsequent analysis by immunoblotting.
Quantitative real-time PCR
Total RNA was isolated from cell lines using TRIZOL reagent (Ambion, #15596026, Carlsbad, CA, USA) in accordance with the manufacturer’s instructions. RNA concentrations were measured using NanoDrop. An amount of 2 μg of total RNA was subjected to DNase treatment using two units of the RQ1 RNase-free DNase (Promega #M6101) for 30 min at 37 °C. Subsequently, cDNA was synthesized using the RevertAid Premium Reverse Transcriptase (Thermo Scientific) kit and oligo(dT) primer. cDNA was diluted 1 : 3 before analysis by qRT-PCR. Small quantities of RNA were extracted and purified using the miRNeasy Micro Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Residual amounts of DNA were removed by on-column DNase digestion. An amount of 500 ng RNA was transcribed into cDNA. Gene expression was quantified by Maxima SYBR Green/Rox qPCR (Thermo Scientific #K0223) performed on a StepOnePlus Real-Time PCR system (Applied Biosystems). Oaz1 and β-Actin as housekeeping genes were used for normalization of loaded cDNA. Gene-specific primers were ordered from Metabion and are listed in Supplementary Table 1.
Immunofluorescence microscopy
For co-localization assays, 105 iLin41 cells were seeded as a monolayer 24 h before processing for immunocytochemistry. Cells were washed twice with ice-cold PBS and fixed with 4% PFA for 10 min at 4 °C. After further washing with PBS, blocking of unspecific binding was performed in 10% FBS, 0.2% gelatin, 0.25% Triton X, 1% BSA in 1 × PBS for 2 h at room temperature. Cells were stained with primary antibodies diluted in blocking solution overnight at 4 °C (see Supplementary Table 2). After washing cells were incubated with 1:500 diluted secondary antibody and optional Draq5 (1:2000) for 1-2 h at room temperature. Alternatively, nuclei were visualized by staining with DAPI during the final washing steps. Finally, the cover slips were mounted to glass slides and co-localization was analyzed using a Leica SL or SP8 confocal microscope.
For preparation of cryo-sections, E9.5 or E10.5 embryos were isolated and fixed in 4% (w/v) paraformaldehyde in PBS overnight and cryo-protected with a 10–20–30% sucrose PBS gradient. On the day of embedding, embryos were incubated in a 30% sucrose/7.5% gelatine solution at 37 °C until sinking. The bottom of the plastic embedding mold was covered with the sucrose/gelatine solution and incubated at 4 °C for 1 h. On top of this solidified layer, the embryo was then placed in the desired orientation according the sectioning surface and covered avoiding bubbles using the 37 °C pre-warmed gelatin/sucrose solution. In order to quickly freeze the embedding medium, the mold was placed onto a slurry of dry-ice in 2-methylbutane. Before cryo-sectioning, the tissues were stored at −80 °C. Cryo-sections at 12 μm thickness were cut using a Microm HM5000 M cryostat, mounted onto superfrost slides, dried for 1 h at room temperature and stored at −80 °C until use. For immunohistochemistry, frozen slices were thawed at room temperature, profiled with a hydrophobic pen (DAKOpen) and washed with PBS for 20 min under soft shaking. Then sections were pre-incubated with blocking solution (0.25% Triton X, 3% BSA in 1 × PBS) at room temperature for 1 h and incubated with 100 μl primary antibody (for concentration, see Supplementary Table 2) diluted in blocking buffer in a humid chamber at 4 °C overnight. After three 20 min-washing steps with PBS, slices were stained with fluorescence-conjugated secondary antibody protected from light for 2 h. The sections were further washed three times with PBS including DAPI staining in the last washing step. Finally, the sections were mounted with fluorescent-protective mounting media until set. Images were taken using an Olympus BX51 epifluorescence microscope or SP8 confocal microscope and processed with Photoshop or ImageJ.
Cell number and proliferation assay
To compare the proliferation of control and LIN41-induced ES cells, the cell number was monitored over time. Initial seeding was 6.7 × 104 cells per 24-well for pluripotent conditions and 1.3 × 104 cells per 24-well for cells treated with RA. In 24 h intervals, cells were trypsinized for manual counting. Each time point was analyzed in triplicates. Three independent experiments were performed for the statistical analysis.
The Cell Proliferation ELISA Kit (Roche) for colorimetric quantification was used according to the manufacturer’s protocol. Initial seeding was 1 × 104 cells for pluripotent conditions and 2 × 103 cells for cells treated with RA per flat-bottom 96-well. Cells were cultured with or without Dox as appropriate in a final volume of 100 μl. BrdU incorporation was measured using a Varioskan Flash spectral scanning multimode reader (Thermo Scientific) at 370 nm. Each time point was analyzed in triplicate. Three independent experiments were performed for the statistical analysis.
Cell cycle analysis
Cell cycle stages were analyzed under differentiation conditions by staining with propidium iodide (PI).52 A total of 4.8 × 105 cells were seeded per 10 cm dish. After 72 h of RA-induced differentiation, cells were trypsinized. Single-cell suspensions with 2 × 106 cells for each sample were prepared in 0.5 ml ice-cold PBS. For fixing, 4.5 ml cold 70% ethanol was added and samples were incubated for at least 2 h at 4 °C. Cells were pelleted by centrifugation for 5 min at 300 × g and washed with 5 ml PBS. After a further centrifugation step at 1000 × g for 5 min, the pellet was resuspended in 1 ml PI staining buffer (0.1% Triton X-100, 100 μg/ml DNase-free RNase A in PBS). Half of this cell suspension served as PI-negative control. The other half of the sample was stained for 30 min with 10 μg/ml PI (Life Technologies #P3566, Carlsbad, CA, USA) at room temperature in the dark before cell fluorescence of 20 000 events was measured by flow cytometry (BD Canto II). Prior gating for live cells, single cells were gated using width and area parameters of PI. An area parameter histogram was plotted using FlowJo10 software to define percentage of cells in G1, S and G2/M phases.
Statistical analysis
Data collected from at least three independent experiments are presented as mean±S.D. Statistical testing was performed using GraphPad Prism 5. Multi-group comparisons were analyzed by two-way ANOVA with the Bonferroni post-test (Figures 4a, b and Supplementary Figures 3a–c). Statistical differences between two groups were determined by a Student’s t-test. When comparing selected groups with one independent variable one-way ANOVA analysis with Bonferroni’s multiple comparison test was employed. Significance is denoted in the figures as: ***P<0.001; **P<0.01; *P<0.05.
Animal use
All mice experiments were performed according to the applicable European and German laws, following the Animal Welfare Act and the European legislative Directive 86/609/EEC and approved by the Animal Welfare Committee of the Charité, Berlin.
Heterozygote Lin41+/gt mice13 were mated overnight and a positive copulation plug was considered as embryonic Day 0.5 (E0.5). At E9.5 and E10.5, anesthetized mothers were killed by cervical dislocation. Both uterine horns containing embryos were removed and washed in sterile PBS on ice. Each embryo was dissected from the decidua with the yolk sac and amnion and then either immediately fixed in 4% PFA overnight for immunohistochemistry or flash-frozen in liquid nitrogen for RNA and protein extraction. For genotyping, genomic DNA was extracted from the yolk sac by lysis in 100 μl DirectPCR Lyse Reagent Tail (Peqlab 31-101-T) and 0.3 mg/ml Proteinase K for 30 min at 55 °C. Subsequently, Proteinase K was inactivated by heating for 45 min at 85 °C. After centrifugation for 10 s at maximum speed to pellet tissue debris, 1 μl of the supernatant was subjected to genotyping. PCR was performed using Go-Taq DNA polymerase kit (Promega #M3008) and three primers (Exon 2 F, Exon 2 R and GT R) to discriminate between the wild type (457 bp) and gene trap (600 bp) allele (for primer sequence, see Supplementary Table 1).
References
Strikoudis A, Guillamot M, Aifantis I . Regulation of stem cell function by protein ubiquitylation. EMBO Rep 2014; 15: 365–382.
Slack FJ, Basson M, Liu Z, Ambros V, Horvitz HR, Ruvkun G . The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol Cell 2000; 5: 659–669.
Ecsedi M, Rausch M, Grosshans H . The let-7 microRNA directs vulval development through a single target. Dev Cell 2015; 32: 335–344.
Worringer KA, Rand TA, Hayashi Y, Sami S, Takahashi K, Tanabe K et al. The let-7/LIN-41 pathway regulates reprogramming to human induced pluripotent stem cells by controlling expression of prodifferentiation genes. Cell Stem Cell 2014; 14: 40–52.
Chang H-M, Martinez NJ, Thornton JE, Hagan JP, Nguyen KD, Gregory RI . Trim71 cooperates with microRNAs to repress Cdkn1a expression and promote embryonic stem cell proliferation. Nat Commun 2012; 3: 923.
Kwon SC, Yi H, Eichelbaum K, Föhr S, Fischer B, You KT et al. The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol 2013; 20: 1122–1130.
Loedige I, Gaidatzis D, Sack R, Meister G, Filipowicz W . The mammalian TRIM-NHL protein TRIM71/LIN-41 is a repressor of mRNA function. Nucleic Acids Res 2013; 41: 518–532.
Loedige I, Jakob L, Treiber T, Ray D, Stotz M, Treiber N et al. The crystal structure of the NHL domain in complex with RNA reveals the molecular basis of drosophila brain-tumor-mediated gene regulation. Cell Rep 2015; 13: 1206–1220.
Rybak A, Fuchs H, Hadian K, Smirnova L, Wulczyn EA, Michel G et al. The let-7 target gene mouse lin-41 is a stem cell specific E3 ubiquitin ligase for the miRNA pathway protein Ago2. Nat Cell Biol 2009; 11: 1411–1420.
Chen J, Lai F, Niswander L . The ubiquitin ligase mLin41 temporally promotes neural progenitor cell maintenance through FGF signaling. Genes Dev 2012; 26: 803–815.
Wulczyn FG, Cuevas E, Franzoni E, Rybak A . miRNAs need a trim: regulation of miRNA activity by Trim-NHL proteins. AdvExpMedBiol 2011; 700: 85–105.
Maller Schulman BR, Liang X, Stahlhut C, DelConte C, Stefani G, Slack FJ . The let-7 microRNA target gene, Mlin41/Trim71 is required for mouse embryonic survival and neural tube closure. Cell Cycle 2008; 7: 3935–3942.
Cuevas E, Rybak-Wolf A, Rohde AM, Nguyen DTT, Wulczyn FG . Lin41/Trim71 is essential for mouse development and specifically expressed in postnatal ependymal cells of the brain. Front Cell Dev Biol 2015; 3: 20.
Mitschka S, Ulas T, Goller T, Schneider K, Egert A, Mertens J et al. Co-existence of intact stemness and priming of neural differentiation programs in mES cells lacking Trim71. Sci Rep 2015; 5: 11126.
Rinon A, Molchadsky A, Nathan E, Yovel G, Rotter V, Sarig R et al. p53 coordinates cranial neural crest cell growth and epithelial-mesenchymal transition/delamination processes. Development 2011; 138: 1827–1838.
Montes de Oca Luna R, Wagner DS, Lozano G . Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995; 378: 203–206.
Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, Jacks T . A subset of p53-deficient embryos exhibit exencephaly. Nat Genet 1995; 10: 175–180.
Armstrong JF, Kaufman MH, Harrison DJ, Clarke AR . High-frequency developmental abnormalities in P53-deficient mice. Curr Biol 1995; 5: 931–936.
Eiraku M, Sasai Y . Mouse embryonic stem cell culture for generation of three-dimensional retinal and cortical tissues. Nat Protoc 2012; 7: 69–79.
Han M-K, Song E-K, Guo Y, Ou X, Mantel C, Broxmeyer HE . SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell 2008; 2: 241–251.
Aladjem MI, Spike BT, Rodewald LW, Hope TJ, Klemm M, Jaenisch R et al. ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr Biol 1998; 8: 145–155.
Shigeta M, Ohtsuka S, Nishikawa-Torikai S, Yamane M, Fujii S, Murakami K et al. Maintenance of pluripotency in mouse ES cells without Trp53. Sci Rep 2013; 3: 2944.
Meletis K, Wirta V, Hede S-M, Nistér M, Lundeberg J, Frisén J . p53 suppresses the self-renewal of adult neural stem cells. Development 2006; 133: 363–369.
Gil-Perotin S, Marin-Husstege M, Li J, Soriano-Navarro M, Zindy F, Roussel MF et al. Loss of p53 induces changes in the behavior of subventricular zone cells: implication for the genesis of glial tumors. J Neurosci 2006; 26: 1107–1116.
Solozobova V, Blattner C . p53 in stem cells. World J Biol Chem 2011; 2: 202–214.
Migliorini D, Lazzerini Denchi E, Danovi D, Jochemsen A, Capillo M, Gobbi A et al. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol 2002; 22: 5527–5538.
Van Nostrand JL, Brady CA, Jung H, Fuentes DR, Kozak MM, Johnson TM et al. Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature 2014; 514: 228–232.
Brynczka C, Labhart P, Merrick BA . NGF-mediated transcriptional targets of p53 in PC12 neuronal differentiation. BMC Genomics 2007; 8: 139–17.
Ting SB, Wilanowski T, Auden A, Hall M, Voss AK, Thomas T et al. Inositol- and folate-resistant neural tube defects in mice lacking the epithelial-specific factor Grhl-3. Nat Med 2003; 9: 1513–1519.
Kimura-Yoshida C, Mochida K, Ellwanger K, Niehrs C, Matsuo I . Fate specification of neural plate border by canonical Wnt signaling and Grhl3 is crucial for neural tube closure. EBioMedicine 2015; 2: 513–527.
Nikolopoulou E, Galea GL, Rolo A, Greene NDE, Copp AJ . Neural tube closure: cellular, molecular and biomechanical mechanisms. Development 2017; 144: 552–566.
Cenci C, Gould AP . Drosophila grainyhead specifies late programmes of neural proliferation by regulating the mitotic activity and Hox-dependent apoptosis of neuroblasts. Development 2005; 132: 3835–3845.
Cheung CC, Yang C, Berger T, Zaugg K, Reilly P, Elia AJ et al. Identification of BERP (brain-expressed RING finger protein) as a p53 target gene that modulates seizure susceptibility through interacting with GABAA receptors. Proc Natl Acad Sci USA 2010; 107: 11883–11888.
Raheja R, Liu Y, Hukkelhoven E, Yeh N, Koff A . The ability of TRIM3 to induce growth arrest depends on RING-dependent E3 ligase activity. Biochem J 2014; 458: 537–545.
Liu J, Zhang C, Wang XL, Ly P, Belyi V, Xu-Monette ZY et al. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ 2014; 21: 1792–1804.
Pant V, Lozano G . Limiting the power of p53 through the ubiquitin proteasome pathway. Genes Dev 2014; 28: 1739–1751.
Regeling A, Armata HL, Gallant J, Jones SN, Sluss HK . Mice defective in p53 nuclear localization signal 1 exhibit exencephaly. Transgenic Res 2010; 20: 899–912.
Marchenko ND, Hanel W, Li D, Becker K, Reich N, Moll UM . Stress-mediated nuclear stabilization of p53 is regulated by ubiquitination and importin-alpha3 binding. Cell Death Differ 2010; 17: 255–267.
Brooks CL, Gu W . The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011; 2: 456–462.
Lee D-F, Su J, Ang Y-S, Carvajal-Vergara X, Mulero-Navarro S, Pereira CF et al. Regulation of embryonic and induced pluripotency by aurora kinase-p53 signaling. Cell Stem Cell 2012; 11: 179–194.
Wang Q, Zou Y, Nowotschin S, Kim SY, Li QV, Soh C-L et al. The p53 family coordinates Wnt and nodal inputs in mesendodermal differentiation of embryonic stem cells. Cell Stem Cell 2017; 20: 70–86.
Quadrato G, Di Giovanni S . Gatekeeper between quiescence and differentiation: p53 in axonal outgrowth and neurogenesis. Int Rev Neurobiol 2012; 105: 71–89.
Copp AJ, Greene NDE, Murdoch JN . The genetic basis of mammalian neurulation. Nat Rev Genet 2003; 4: 784–793.
Yamaguchi Y, Miura M . Programmed cell death in neurodevelopment. Dev Cell 2015; 32: 478–490.
Duan WR, Garner DS, Williams SD, Funckes-Shippy CL, Spath IS, Blomme EAG . Comparison of immunohistochemistry for activated caspase-3 and cleaved cytokeratin 18 with the TUNEL method for quantification of apoptosis in histological sections of PC-3 subcutaneous xenografts. J Pathol 2003; 199: 221–228.
Rifat Y, Parekh V, Wilanowski T, Hislop NR, Auden A, Ting SB et al. Regional neural tube closure defined by the Grainy head-like transcription factors. Dev Biol 2010; 345: 237–245.
Melton C, Judson RL, Blelloch R . Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature 2010; 463: 621–626.
Kamiya D, Banno S, Sasai N, Ohgushi M, Inomata H, Watanabe K et al. Intrinsic transition of embryonic stem-cell differentiation into neural progenitors. Nature 2011; 470: 503–509.
Lundgren DH, Martinez H, Wright ME, Han DK . Protein identification using Sorcerer 2 and SEQUEST. Curr Protoc Bioinformatics 2009; Chapter 13: Unit 13.3–13.3.21.
Huang DW, Sherman BT, Lempicki RA . Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44–57.
Mi H, Muruganujan A, Casagrande JT, Thomas PD . Large-scale gene function analysis with the PANTHER classification system. Nat Protoc 2013; 8: 1551–1566.
Pozarowski P, Darzynkiewicz Z . Analysis of cell cycle by flow cytometry. Methods Mol Biol 2004; 281: 301–311.
Acknowledgements
We thank Richard Gregory (iLin41) and Hitoshi Niwa (p53 knockout) for providing ES cell lines. We are grateful to Frederick Rehfeld, Claudia Marini and Pina Knauff for critically reading the manuscript, all current and former members of the Wulczyn lab especially Agnieszka Rybak-Wolf and F Rehfeld for helpful discussions and Annette Hammes-Lewin for advice on imaging mouse embryos. We thank Jutta Schüler and Jörg Rösner from the microscope facility for their technical support. The Leica TCS SP8 was funded by BMWi/DLR (Grant 50WB1421), Center of Space Medicine Berlin. Byron Ricketts and Jeffrey Silva coordinated our collaboration with Cell Signaling Technologies. This work was funded by the Berliner Krebsgesellschaft e.V. and the DFG Collaborative Research Program 665: Developmental Disturbances in the Nervous System.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Nguyen, D., Richter, D., Michel, G. et al. The ubiquitin ligase LIN41/TRIM71 targets p53 to antagonize cell death and differentiation pathways during stem cell differentiation. Cell Death Differ 24, 1063–1078 (2017). https://doi.org/10.1038/cdd.2017.54
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2017.54
This article is cited by
-
TRIM family contribute to tumorigenesis, cancer development, and drug resistance
Experimental Hematology & Oncology (2022)
-
Impaired neurogenesis alters brain biomechanics in a neuroprogenitor-based genetic subtype of congenital hydrocephalus
Nature Neuroscience (2022)
-
p53 induces ARTS to promote mitochondrial apoptosis
Cell Death & Disease (2021)
-
Role of p53 deficiency in socket healing after tooth extractions
Journal of Molecular Histology (2020)
-
CSB affected on the sensitivity of lung cancer cells to platinum-based drugs through the global decrease of let-7 and miR-29
BMC Cancer (2019)
