
Abstract
Recent studies have suggested that C-MYC may be an excellent therapeutic cancer target and a number of new agents targeting C-MYC are in preclinical development. Given most therapeutic regimes would combine C-MYC inhibition with genotoxic damage, it is important to assess the importance of C-MYC function for DNA damage signalling in vivo. In this study, we have conditionally deleted the c-Myc gene in the adult murine intestine and investigated the apoptotic response of intestinal enterocytes to DNA damage. Remarkably, c-Myc deletion completely abrogated the immediate wave of apoptosis following both ionizing irradiation and cisplatin treatment, recapitulating the phenotype of p53 deficiency in the intestine. Consistent with this, c-Myc-deficient intestinal enterocytes did not upregulate p53. Mechanistically, this was linked to an upregulation of the E3 Ubiquitin ligase Mdm2, which targets p53 for degradation in c-Myc-deficient intestinal enterocytes. Further, low level overexpression of c-Myc, which does not impact on basal levels of apoptosis, elicited sustained apoptosis in response to DNA damage, suggesting c-Myc activity acts as a crucial cell survival rheostat following DNA damage. We also identify the importance of MYC during DNA damage-induced apoptosis in several other tissues, including the thymus and spleen, using systemic deletion of c-Myc throughout the adult mouse. Together, we have elucidated for the first time in vivo an essential role for endogenous c-Myc in signalling DNA damage-induced apoptosis through the control of the p53 tumour suppressor protein.
Similar content being viewed by others


Main
A well-known function of the c-Myc protein is its ability to drive apoptosis in numerous cellular contexts.1, 2, 3 Of the studies performed, most have concentrated on the ability of c-Myc overexpression to drive apoptosis, unless accompanied by other mutations such as p53 loss.4 The suppression of apoptosis is thought to be a key factor in driving tumorigenesis in vivo, for example overexpression of c-Myc in pancreatic islets alone does not induce tumorigenesis unless apoptosis is blocked, for example, by p53 loss, Bcl-xl overexpression or ARF knockout.5, 6 The studies examining combined c-Myc overexpression and p53 loss have implicated p53 directly downstream of c-Myc, but whether this is a direct transcriptional control or indirect (e.g., through c-Myc induction of the DNA damage response) is still controversial.4 The most cited model linking c-Myc overexpression to the p53 pathway is via transcription induction of ARF by c-Myc, which in turn inhibits Mdm2 (a key negative regulator of p53).7, 8 Indeed, in mouse, overexpressing high levels of c-Myc-ER from the Rosa26 locus showed that c-Myc induced apoptosis only in the colon. This was due to higher expression of the Rosa26 locus and hence overexpression of c-Myc-ER within the colon compared with other tissues, leading to the induction of ARF and apoptosis. Genetic deletion of Arf rescued this c-Myc-induced apoptosis.9
The importance of c-Myc in signalling apoptosis following DNA damage is poorly understood. Thus far, no study has examined this in vivo, although in vitro studies have suggested it may be of vital importance. There are a number of lines of evidence for this; first (and most importantly) Seoane et al.10 have shown that in colorectal cancer cell lines depletion of C-MYC reduces apoptosis as a consequence of altering the balance of downstream effectors of P53 signalling. Thus, in the absence of C-MYC, there are increased levels of the antiapoptotic cell cycle arrest protein P21 (a target of P53 which is also transcriptional repressed by C-MYC in a complex with MIZ) and reduced levels of pro-apoptotic genes such as BAX, resulting in cell cycle arrest rather than apoptosis. Second, numerous C-MYC transcriptional targets (either activated or repressed by C-MYC) such as BAX, GADD45A and ONZIN have been shown to be crucial for DNA damage signalling in vitro.11, 12, 13 Third, c-Myc has been shown to augment apoptosis in fibroblasts following gamma irradiation.14 Finally, the post transcriptional regulation of c-Myc through mir34 family members has been suggested to directly impinge on c-Myc function and the cells response to cytotoxic agents.15
With the promise of emerging cancer therapies to target C-MYC in vivo for the first time using BET inhibitors, there is an urgent requirement to fully understand the role of C-MYC in response to DNA damage. Studies by the Evan group have shown that inhibiting endogenous murine c-Myc in vivo through an inducible dominant negative c-Myc protein (OMOMYC) causes regression of a variety of murine tumours (including lung tumours induced by Kras mutation and pancreatic neuroendocrine tumours in mice carrying the Riptag transgene).16, 17 Moreover, the effects of c-Myc loss were well-tolerated suggesting that c-Myc is an attractive therapeutic target. These data were consistent with our previous studies demonstrating that c-Myc was absolutely required for the phenotypes associated with deletion of Apc, although the normal intestine could proliferate without c-Myc (albeit at lower levels).18
One of the most tractable systems for studying the DNA damage response in vivo is the intestinal crypt. Previously, numerous cytotoxic agents such as cisplatin, ionizing radiation and N-methyl-N-nitrosourea have been shown to induce apoptosis with a peak induction normally 6–12 h following DNA damage.19, 20 This early wave of apoptosis is completely dependent on the nuclear accumulation of p53. The tractability of this system in conjunction with the our previous data showing that c-Myc deletion is not immediately deleterious to intestinal enterocytes makes this an ideal system to determine whether c-Myc is important for signalling apoptosis in normal cells following DNA damage.18 Importantly, neither of the two studies that conditionally deleted c-Myc from the normal intestine has seen any changes in the physiological levels of apoptosis, which could have possibly confounded any analysis.18, 21 Both studies showed that c-Myc-deficient enterocytes could proliferate; however, our study showed that both the level of proliferation and cell size were reduced compared with wild-type intestinal enterocytes.
In this study, we show that c-Myc is essential for the induction of apoptosis within the intestinal crypt due to the inability of c-Myc-deficient cells to efficiently upregulate p53. Mechanistically, this is associated with high levels of Mdm2 in MYC-deficient cells, and treatment with the MDM2 inhibitor Nutlin restored the stabilization of p53 and induced apoptosis. Importantly, subtle deregulation of c-Myc also has a marked impact on the apoptotic response following DNA damage but no effect on the normal intestine. We also find that other radio-sensitive tissues show a dependence on c-Myc for DNA damage-induced apoptosis. Thus, we propose a general requirement for c-Myc expression in making cells permissive to DNA damage-induced apoptosis in vivo.
Results
c-Myc-deficient crypts do not undergo apoptosis following treatment with DNA-damaging agents
We first examined whether c-Myc deletion could alter the DNA damage response to ionizing radiation. To induce Cre-mediated gene deletion, AhCre+ Mycfl/fl mice and control AhCre+ Myc+/+ mice were given three injections IP of 80 mg/kg β-naphthoflavone within a single day. This protocol leads to near constitutive levels of c-Myc deletion from the intestinal epithelium 4 days following Cre induction18 (Supplementary Figure 1a). At this stage, no Cre recombinase expression can be detected (gene loss remains as the deletion event occurs within the stem cell population at the base of the crypts).22 AhCre+ Mycfl/fl mice and control AhCre+ Myc+/+ mice were then exposed to 14 Gray (Gy) of gamma irradiation, and the induction of apoptosis was scored 6 h following the irradiation.
As has previously been reported, H&E analysis of wild-type mice showed a clear induction of apoptosis following 14 Gy gamma irradiation compared with their nonirradiated littermates (Figures 1a and b).19, 20 However, the number of apoptotic figures was significantly reduced in c-Myc-deficient crypts given the same dose of irradiation (Figures 1a and b). Indeed, the level of apoptosis in these crypts was closer to nonirradiated controls than irradiated wild-type crypts (Figure 1b). The reduction in apoptosis was not explained due to c-Myc-deficient crypts having fewer cells than wild type,18 as we also observed a decrease in the percentage of crypt cells undergoing apoptosis upon c-Myc deletion (Supplementary Figure 1b). To confirm the scoring of apoptosis on H&E sections, immunohistochemistry (IHC) was performed against cleaved (‘active’) caspase 3,23 and once again the number of caspase 3-positive cells was significantly lower in irradiated c-Myc-deficient crypts when compared with wild-type mice given the same dose of gamma irradiation (14 Gy) (Figures 1c and d). To extend this analysis beyond a single time point, we next scored apoptosis at a series of different times following gamma irradiation and found significantly lower levels of apoptosis at all time points subsequent to 2 h in c-Myc-deficient intestinal crypts (Figure 1e).

MYC-deficient crypts do not undergo apoptosis following treatment with DNA-damaging agents. (a) H&E staining of wild type (AhCre+ Myc+/+) and MYC deficient (AhCre+ Mycfl/fl) intestines 6 h following 14 Gy irradiation, arrows show apoptotic figures in wild-type mice. Scale bars=50 μm. (b) Scoring of apoptotic figures from H&E sections shows a significant decrease in apoptosis in MYC-deficient mice following 14 Gy irradiation compared with wild type (* Wt versus Wt + 14 Gy, P =0.04, Mann Whitney n=3 versus 6, ** Wt + 14 Gy versus Myc + 14 Gy, P =0.0041, Mann Whitney n=6 versus 5). (c) Immunohistochemical staining for cleaved (‘active’) Caspase 3 was performed on intestinal sections of wild type and MYC-deficient mice. Scale bars=50 μm. (d) Quantification of these sections revealed a significant decrease in the number of Caspase-3-positive cells in MYC-deficient mice following 14 Gy irradiation compared with wild type (* Wt versus Wt +14 Gy and Wt + 14 Gy versus Myc + 14 Gy, P=0.04, Mann Whitney n=3) (e) Graph showing that MYC is essential for the induction of apoptosis following 14 Gy irradiation. Each time point represents at least three mice, illustrating significantly lower levels of apoptosis in MYC-deficient mice at all time points after 2 h (* wt versus Myc, P=0.04, Mann Whitney n=3. Error bars are S.D.). (f) Scoring of apoptotic figures from H&E sections shows a significant decrease in apoptosis in MYC-deficient mice compared with wild type following lower levels of irradiation (5 Gy) (* wt versus Myc, P=0.04, Mann Whitney n=3). (g) Scoring of apoptotic figures from H&E sections shows a significant decrease in apoptosis in MYC-deficient mice following 10 mg/kg cisplatin treatment compared with wild type (* wt versus Myc, P=0.04, Mann Whitney n=3)
The resistance to apoptosis of c-Myc-deficient enterocytes was not restricted to high doses of γ irradiation, as apoptosis following either a lower dose of irradiation (5 Gy) or cisplatin treatment was also found to be c-Myc dependent (Figures 1f and g). This failure to undergo apoptosis was not simply because c-Myc-deficient cells were not cycling, as we and others have previously shown that c-Myc-deficient intestinal enterocytes can undergo proliferation.18
c-Myc-deficient enterocytes do not upregulate p53
The phenotype of c-Myc deletion paralleled the well-established phenotype of p53 deficiency in the intestine, namely a strong suppression of the immediate wave of apoptosis.20, 24, 25 Taking this information with previous data suggesting direct links between c-Myc and p53, we investigated the kinetics of p53 induction in the murine small intestine.3 Using immunohistochemistry and western blot, we observed a sharp rise in p53 levels following gamma irradiation that is attenuated in c-Myc-deficient crypts (Figures 2a and b and Supplementary Figure 2a). Therefore, this provided a ready mechanism for the abrogated apoptosis.

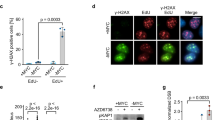
Myc deletion prevents P53 accumulation after DNA damage. (a) IHC for P53 or P21 in wildtype (AhCre Myc+/+) or MYC-deficient (AhCre Mycfl/f) mice 6 h after 14 Gy γ-irradiation. Note the induction of nuclear P53 following irradiation in wild-type mice that is attenuated in MYC-deficient enterocytes, and the upregulation of P21 following irradiation in both wild type and MYC-deficient enterocytes. Scale bars=50 μm. (b) Immunoblotting shows a marked increase in P53 levels following irradiation that is not seen in MYC-deficient intestinal extracts (top panels). Immunoblotting shows a marked increase in P21 levels following irradiation in both wild type and MYC-deficient intestinal extracts (bottom panels). (c) Scoring of apoptotic cells per crypt on the genotypes indicated 6 h after exposure to 14 Gy. AhCre+ Mycfl/fl P21−/− mice display the same lack of apoptotic response to 14 Gy irradiation as the MYC-deficient mice, illustrating that the induction of P21 in AhCre Mycfl/f mice is not responsible for the failure to upregulate P53 and induce apoptosis (AhCre Mycfl/fl versus AhCre+ Mycfl/fl P21−/−, P=0.7656, Mann Whitney n=5 versus 3). (d) Immunohistochemistry for γ-H2AX in wild-type (AhCre Myc+/+) or MYC-deficient (AhCre Mycfl/f) mice following 14 Gy γ-irradiation or cisplatin treatment. The large upregulation of γ-H2AX 30 min after irradiation is beginning to clear by 6 h after irradiation as DNA damage is repaired. This expression pattern is observed in both wild type and MYC-deficient crypts (and cisplatin treated), illustrating that MYC-deficient enterocytes are able to sense DNA damage stimuli. Scale bars=50 μm
An alternative mechanism for failed apoptosis is that deletion of c-Myc causes a derepression of p21, which could also block apoptosis in this system.10, 26 We and others have previously shown that c-Myc deficiency alone is not sufficient to trigger p21 upregulation in the intestine, however, following combined Apc and c-Myc deletion a clear induction of p21 was observed.18, 27, 28 Following irradiation (despite the reduced p53 activation), p21 was still upregulated in c-Myc-deficient intestinal enterocytes (Figures 2a and b and Supplementary Figure 2b). To determine whether the lack of transcriptional repression of p21 by c-Myc was sufficient to block apoptosis, we intercrossed p21 knockout mice to mice carrying the Ah Cre transgene and loxP-flanked Myc alleles to generate AhCre+ Mycfl/fl P21−/− mice. Cre was induced in these mice as described above, and these mice were irradiated 4 days after Cre induction and apoptosis was scored 6 h following 14 Gy irradiation. Importantly, AhCre+ Mycfl/fl P21−/− mice displayed the same lack of apoptosis in response to irradiation as the single c-Myc-deficient intestinal crypts (Figure 2c). Therefore, this demonstrates that the induction of p21 following gamma irradiation was not responsible for the block of apoptosis in c-Myc-deficient enterocytes and that reduced p53 upregulation was the most likely cause of abrogated apoptosis.
c-Myc-deficient enterocytes sense the DNA damage stimuli
From the literature, there are numerous potential mechanisms that could explain the failure to see increased levels of p53 protein in c-Myc-deficient enterocytes. These include failure to detect DNA damage, reduced p53 protein stability or reduced transcription or translation of p53 mRNA.3 Therefore, we decided to test a number of these potential mechanisms.
First, we investigated whether DNA damage recognition was functioning in c-Myc-deficient intestinal enterocytes and examined whether there was efficient activation of the DNA damage sensing proteins γH2AX and ATM. This is particularly important as it has previously been suggested that ATM is required for c-Myc to activate p53.29 H2AX becomes phosphorylated by ATM at the sites of double-strand breaks in DNA and is essential for their recognition and repair.30 We performed immunohistochemistry using an antibody that specifically recognises the activated, phosphorylated form of H2AX (γH2AX) to determine whether this DNA damage response was still intact in c-Myc-deficient mice. In wild-type mice, the level of γH2AX is markedly increased 30 min after irradiation, and this level decreases 6 h later as DNA damage is repaired. The activation of γH2AX following either γ-irradiation or cisplatin was also observed in MYC-deficient intestinal enterocytes (Figure 2d). We also used an antibody specific to the activated, serine 1981 phosphorylated form of ATM. ATM is a regulator of cellular response to DNA damage, and is auto phosphorylated and associates with other proteins such as p53, Mdm2 and Chk2 to arrest cell cycle at G1. In common with the γH2AX results, ATM is still phosphorylated on serine 1981 in response to DNA damage in wild-type and c-Myc-deficient mice following irradiation or cisplatin treatment (Supplementary Figure 2c). Moreover, serine 345 phosphorylation of Chk1 that occurs downstream of ATM and Chk2 was observed at equivalent levels in wild-type and c-Myc-deficient intestines following irradiation or cisplatin treatment (Supplementary Figure 2d). Taken together, these results demonstrate that the DNA damage response is still intact in c-Myc-deficient enterocytes and suggests that the mechanism behind the failure to induce apoptosis in response to DNA damage is through the control of p53 levels.
Mdm2 upregulation in c-Myc-deficient enterocytes stops p53 accumulation and apoptosis following DNA damage
Given the number of studies in the literature that link c-Myc to control of p53 stability, we next investigated the Mdm2-p53 pathway.7, 8 One of the key regulators of p53 protein stability is the Mdm2 E3 ubiquitin ligase. Loss of Mdm2 in vivo leads to embryonic death due to the activation of high levels of p53, which can be rescued by co-deletion of p53.31, 32
First we examined the levels of Mdm2 following c-Myc deletion by immunohistochemistry and immunoblotting. We found a marked upregulation of Mdm in c-Myc-deficient cells that was maintained following irradiation (Figures 3a and b). To test whether this was functionally important for blocking apoptosis in the c-Myc-deficient crypt cells, we employed the Mdm2 antagonist, Nutlin-3a. Nutlin is a selective small-molecule inhibitor of the p53-Mdm2 interaction that releases p53 from Mdm2 control, leading to accumulation of the tumour suppressor protein and activation of the P53 pathway.33, 34 Treatment of cancer cells with wild-type P53 induces cell arrest and apoptosis in vitro and suppresses the growth of human tumour xenografts in nude mice.33, 35

Myc deletion causes an accumulation of MDM2. (a) MDM2 IHC showing no expression in intestinal crypts of wild type (AhCre Myc+/+) and a large increase of MDM2 expression in MYC-deficient (AhCre Mycfl/fl) mice in both nonirradiated and 14 Gy irradiated mice. Scale bars=25 μm. (b) Immunoblotting for MDM2 shows a marked increase in MDM2 protein levels in both nonirradiated and 14 Gy irradiated mice MYC-deficient intestines. (c) MYC-deficient mice treated with Nutlin exhibited a full restoration of the apoptotic response (* wt versus Myc, P =0.04, Mann Whitney n=3), which was not observed in MYC-deficient mice treated with vehicle. Note this restoration of apoptosis correlates with an induction of P53 in MYC-deficient mice treated with Nutlin. (d) Immunohistochemistry for P53 demonstrating that P53 now accumulates in nutlin-treated MYC-deficient intestines following gamma irradiation but not vehicle-treated or nonirradiated MYC-deficient intestines. Scale bars=50 μm
c-Myc-deficient mice were treated with Nutlin twice daily on days 1–3 post Cre induction and a final time 3 h before 14 Gy irradiation on day 4. This methodology was employed as it has been shown previously that Nutlin can knockdown Mdm2 for approximately 12 h in vivo, and a number of doses are required for full functional inhibition.33 Apoptosis was scored in WT and c-Myc-deficient mice 6 h following irradiation and a restoration of the apoptotic response was observed in c-Myc-deficient mice treated with Nutlin, though not vehicle (Figure 3c). Consistent with previous reports, Nutlin treatment had no obvious impact on proliferation or apoptosis of wild-type intestinal enterocytes (either alone or treated with 14 Gy) (data not shown).35 Most importantly, this restoration of the apoptotic response correlated with the induction of p53 in the c-Myc-deficient mice treated with Nutlin (Figure 3d). These data demonstrate that restoration of p53 function, via Nutlin treatment, is sufficient to rescue the blocked apoptosis phenotype in c-Myc-deficient enterocytes and therefore establishes that c-Myc regulates apoptosis via p53 in the mammalian intestine.
Deregulated overexpression of c-Myc increases and sustains DNA damage-induced apoptosis
Our data demonstrate that MYC loss of function completely abrogates the apoptotic response induced by DNA damage. We next asked if subtle overexpression of c-Myc could influence the DNA damage response. To alter c-Myc levels, we used mice where the c-Myc cDNA has been targeted in the ROSA26 locus and is under the control of Cre expression through Lox Stop Lox elements in the promoter (ROSA-Floxed-Stop (RFS)-mycWT).36 This system permits inducible, relatively low level, deregulated c-Myc overexpression, previously shown to be sufficient to overcome the reduced proliferation in c-Myc-deficient intestines.37
Following activation of c-Myc transgene expression, we induced DNA damage using 5 Gy IR and examined the impact of increasing levels of c-Myc for the apoptotic response. This lower level of DNA damage permits observations over extended time points. Increasing c-Myc levels had no impact on the basal levels of apoptosis (Figures 4a and b, 0 h time point). Following irradiation, however, the peak apoptotic response at 6 h was significantly increased (Figures 4a and b). In addition, whereas in control mice the rate of apoptosis had dropped sharply 24 h after DNA damage, it was maintained at a significantly higher level in intestines expressing deregulated c-Myc (Figures 4a and b). To confirm these observations, immunohistochemistry against active caspase 3 was performed. In line with the histological data, increased c-Myc levels led to increased numbers of caspase 3-positive cells (Figures 4a and c). Thus, in addition to being required for intestinal apoptosis following DNA damage, deregulated c-Myc expression can also elevate and maintain it. These data strongly suggest that the precise regulation of c-Myc levels is important for the kinetics of apoptosis following DNA damage.

Deregulated MYC expression increases DNA damage-induced apoptotic response. (a) H&E staining (top panels) and caspase 3 IHC (bottom panels) of wild type (AhCre Rosa26+/+) and Myc transgene expressing (RFS-mycWT) small intestines 0, 6, 24 and 48 h following 5 Gy irradiation, arrows indicate apoptotic bodies and caspase 3-positive cells. Scale bars=50 μm. (b) Scoring of apoptotic bodies from H&E sections shows a significant increase in apoptosis in small intestines overexpressing MYC at 6 h (* WT versus RFS-mycWT, P=0.0184, Mann Whitney n=5 versus 3) and 24 h (* WT versus RFS-mycWT, P=0.04, Mann Whitney n=3) following 5 Gy irradiation (Error bars are standard deviation). (c) Scoring of caspase 3-positive cells shows a significant increase in apoptosis in small intestines overexpressing MYC at 24 h (* WT versus RFS-mycWT, P=0.04, Mann Whitney n=3) and 48 h (* WT versus RFS-mycWT, P=0.04, Mann Whitney n=3) following 5 Gy irradiation (Error bars are S.D.)
Loss of c-Myc throughout the adult mouse prevents DNA damage-induced apoptosis in radiosensitive tissues
Given therapeutic inhibition of C-MYC would likely suppress its function throughout the body, we next tested if our findings extended beyond the intestinal epithelium. Outside of the small intestine other radiosensitive tissues include the colon, spleen and thymus. Importantly, the adult thymus proliferates at a very low rate, and thus this is an excellent tissue to study the induction of p53-dependent apoptosis that is uncoupled from proliferation. The ubiquitously expressed RosaCreER permits conditional gene deletion in multiple tissues. To test recombination rates, we generated RosaCreER+ Lox-stop-lox RFP mice and induced them with tamoxifen. High levels of RFP expression were observed in multiple tissues including the colon, spleen and thymus indicating successful recombination in these tissues (Supplementary Figures 3a–c). To test if c-Myc is a critical mediator of apoptosis in these tissues, we induced control RosaCreER+ Myc+/+ and experimental RosaCreER+ Mycfl/fl mice, irradiated them with 14 Gy 6 days post induction and killed them 6 h later. In the colonic epithelium, we observed significantly decreased levels of apoptosis (Figures 5a and b) that was coincident with loss of c-Myc protein (Supplementary Figure 3d). As before, we confirmed this observation with caspase 3 staining (Figures 5c and d). Scoring of caspase 3 positivity also indicated a significantly reduced apoptotic response in both the spleen and thymus (Figures 5e and h). Similar to the small intestine, we observed attenuated p53 induction in the spleen and thymus indicating that c-Myc-dependent upregulation of p53 may be responsible for the induction of apoptosis in multiple tissues. (Supplementary Figure 4a and b). Interestingly, despite a failure to induce apoptosis, p53 induction was comparable between WT and c-Myc-deficient colonic cells (Supplementary Figure 4c). This indicates that, in the colon, c-Myc either controls apoptosis downstream of p53 signalling or via a P53-independent mechanism. Together, these data demonstrate that c-Myc function is required for DNA damage-induced apoptosis in multiple epithelial and lymphoid tissues, strongly suggesting that it is a general mediator of apoptosis in vivo.

MYC deletion prevents DNA damage-induced apoptosis in multiple tissues. (a) H&E staining of wild type (RosaCreERT2 Myc+/+) and MYC-deficient (RosaCreERT2 Mycfl/fl) colons 6 h following 14 Gy irradiation, arrows show apoptotic bodies. Scale bars=50 μm. (b) Scoring of apoptotic bodies from H&E sections shows a significant decrease in apoptosis in MYC-deficient colons following 14 Gy irradiation compared with wild type (** wt versus Myc, P=0.0059, Mann Whitney n=5 versus 7). (c) Caspase 3 IHC staining of wild type (RosaCreERT2 Myc+/+) and MYC-deficient (RosaCreERT2 Mycfl/fl) colons 6 h following 14 Gy irradiation, arrows indicate Caspase 3-positive cells. Scale bars=50 μm. (d) Scoring of Caspase 3-positive cells (percentage of total cells: n>500) shows a significant decrease in apoptotic cells in MYC-deficient colons (** wt versus Myc, P=0.0059, Mann Whitney n=5versus7). (e) Caspase 3 IHC staining of wild type (RosaCreERT2 Myc+/+) and MYC-deficient (RosaCreERT2 Mycfl/fl) spleens 6 h following 14 Gy irradiation, arrows indicate Caspase 3-positive cells. Scale bars=50 μm. (f) Scoring of Caspase 3-positive cells (percentage of total cells: n>500) shows a significant decrease in apoptotic cells in MYC-deficient spleens (* wt versus Myc, P=0.0152, Mann Whitney n=4). (g) Caspase 3 IHC staining of wild type (RosaCreERT2 Myc+/+) and MYC-deficient (RosaCreERT2 Mycfl/fl) thymus 6 h following 14 Gy irradiation, arrows indicate Caspase 3-positive cells. Scale bars=50 μm. (h) Scoring of Caspase 3-positive cells (percentage of total cells: n>500) shows a significant decrease in apoptotic cells in MYC-deficient thymus (* wt versus Myc, P=0.0152, Mann Whitney n=4)
Discussion
Here, we demonstrate for the first time in an in vivo setting that endogenous c-Myc is required for efficient induction of apoptosis following DNA damage. We show that this is due to a failure to upregulate p53 owing to increased levels of the E3 ubiquitin ligase Mdm2 (Figure 6). As this mechanism is conserved in multiple tissues, we contend that c-Myc serves as a universal regulator of apoptosis in vivo.

Model of MYC regulation of apoptosis in response to DNA damage in the intestine. (a) In wild type, MYC-proficient mice, the intestinal epithelial cells respond to DNA damage by upregulating MYC, which then inhibits MDM2. This allows P53 levels to increase and induce apoptosis. (b) When MYC is deleted from the intestinal epithelial cells they can no longer inhibit MDM2 in response to DNA damage, and consequently P53 is not upregulated resulting in reduced apoptosis
c-Myc has the ability to sensitize or induce apoptosis in vitro, but its role in this process is not well established in vivo. We found that deletion of c-Myc throughout the adult mouse strongly suppressed the apoptotic response following DNA damage. c-Myc-deficient intestinal epithelial cells are able to proliferate and thymocytes proliferate very slowly, thus the lack of apoptosis is not simply a byproduct of the proliferative function of c-Myc. Indeed, as overexpression of c-Myc in the small intestine increased both the level and length of the apoptotic response, we believe c-Myc should be regarded as an apoptotic permissivity factor. Thus, it is also likely that the precise regulation of c-Myc throughout DNA damage-induced apoptosis is important for the cessation of the apoptotic response following DNA repair. Interestingly, subtle deregulation of c-Myc expression did not lead to increased levels of intestinal apoptosis in the absence of DNA damage. This appears to contrast with previous studies demonstrating that overexpression of c-Myc-ER leads to ectopic apoptosis in the colon.9 This may be due to a difference in apoptotic response between the small intestine and colon. Alternatively, it may highlight a difference between overexpression of c-Myc-ER and c-Myc. It is possible that c-Myc-ER accumulates to high levels in the absence of tamoxifen. Thus, when tamoxifen is administered, the acute burst of c-Myc-ER activity induced is sufficient to drive apoptosis. As c-Myc expression is regulated by multiple transcriptional and post transcriptional mechanisms in vitro, it is likely that its regulation in vivo is also complex.38 It is interesting to note that previous studies have shown that reducing the c-Myc targeting mir34b/c microRNA causes increased DNA damage apoptosis, suggesting physiological regulation of c-Myc levels following DNA damage modifies the apoptotic response.15
We observed a failure to induce p53 in Myc-deficient intestinal, splenic and thymic cells. In the intestine, this was associated with increased Mdm2 expression and treatment with the Mdm2 inhibitor Nutlin rescued both p53 activation and induction of apoptosis. This suggests that c-Myc regulates apoptosis via post translational regulation of p53 stability in vivo. ATM can also activate p53 suggesting that failed DNA damage signalling could also impact on apoptotic response.29 Importantly, we found DNA damage signalling intact in c-Myc-deficient intestines indicating that control of Mdm2 is the primary mechanism through which c-Myc regulates apoptosis in vivo. The precise mechanism linking c-Myc levels to Mdm2 expression is unclear, but Mdm2 has previously been shown to be a direct transcriptional target of NMYC in neuroblastoma.39 Although this study demonstrated a role in activating Mdm2 transcription, c-Myc proteins are also well-defined transcriptional repressors so it is tempting to speculate that Mdm2 transcription may be directly inhibited by c-Myc. Alternatively, the c-Myc transcriptional target Nucleolin has been shown to directly bind to and inhibit Mdm2.40 This binding was also shown to lead to reduced levels of Mdm2 protein, thus making it an attractive potential mediator of the phenotypes we observe. A thorough dissection of these potential interactions will be important for further understanding of this process.
Interestingly, it does not appear that this mechanism extends to the colon as c-Myc-deficient colonic cells efficiently upregulate P53 following irradiation. This indicates that induction of the apoptotic programme, although c-Myc dependent, is fundamentally different in the colon. With regards to this, the transcriptional activation and oligomerization of the apoptosis regulator Bax has been shown to be controlled by c-Myc.11, 41 Thus, it is possible that colonic cells engage a different apoptotic programme from those in the intestine. Determining the mechanistic basis for this difference will be an interesting future avenue of research.
C-MYC is one of the most commonly altered genes in human cancer with gene amplifications and transcriptional activation especially common. As such, it is a very attractive therapeutic target and several recent studies have highlighted this potential. In particular, c-Myc inhibition using a dominant negative form of the protein termed OMOMYC was shown to regress tumours from both lung and pancreatic mouse tumour models.16, 17 Importantly, although expression of OMOMYC suppressed proliferation in various tissues, these effects were completely reversed upon cessation of treatment. This suggests that C-MYC inhibition would not be overly toxic and is therefore a viable therapeutic target. Inhibition of BET bromodomain proteins has been shown to inhibit C-MYC function and tumorigenesis, thus chemical suppression of MYC may be possible.42 As most C-MYC inhibitory agents would be delivered alongside DNA-damaging agents, our findings that c-Myc is required for apoptosis are important. It will be important to determine if our findings extend to tumorigenic cells as they may also be protected from apoptosis by C-MYC inhibition. If this is the case then precise coordination of treatment regimens may be required to achieve maximum functionality. Interestingly, OMOMYC or BET inhibition both lead to proliferation arrest and apoptosis in transformed cells, indicating that they may not have the same requirement for C-MYC during apoptosis. In fact, this may lead to an unexpected benefit of C-MYC inhibition, namely some protection from the side effects of commonly used therapeutics. It will be important to gain further understanding of this process in vivo to help better understand this issue.
In conclusion, we have demonstrated for the first time, a requirement for c-Myc function during the induction of apoptosis following DNA damage. We find this function is constant in multiple tissues defining c-Myc as a general mediator of apoptosis in vivo.
Materials and Methods
Mouse experiments
All experiments were performed under the UK Home Office guidelines. Outbred male mice from 6 to 12 weeks of age were used, which were segregating for the C57BLJ and S129 genomes. The alleles used were as follows: Mycfl, AhCre, P21, ROSA-Floxed-Stop (RFS)-mycWT, RosaCreER and ROSA-tdRFP.22, 36, 43, 44, 45, 46, 47 Myc experiments were also confirmed on mice that had been backcrossed to C57Bl6J for five generations.
Cre induction was carried out by giving AhCre+ Myc++ and AhCre+Mycfl/fl 3 intraperitoneal (IP) injections of 80 mg/kg β-naphthoflavone in a single day. Mice were then given DNA damage 4 days after induced gene deletion. Previous experiments have shown that, using this protocol, no significant induction of apoptosis is seen in induced (AhCre+ Myc++) when compared with uninduced AhCre+ Myc++ or induced wild type (mice not carrying the AhCre transgene) at day 4 after induction.
For assessing whether MYC deficiency affects the DNA damage response following gamma irradiation ‘wild type’ AhCre+ Myc++ and MYC-deficient AhCre+ Mycfl/fl mice were irradiated with 14 Gy irradiation using a Cs137 source delivered at a dose rate of at 0.423 Gy/min. Mice were then collected at 30 min, 1, 2, 3, 6, 12, 24 and 48 h time points following the irradiation. At least three mice were used for each time point. For cisplatin treatment, mice were given a single IP injection of 10 mg/kg Cisplatin (purchased from David Bull Laboratories (Warwick, UK) and distributed by Faulding Pharmaceuticals).
For assessing whether MDM2 upregulation abrogated apoptosis following Myc deletion, ‘wild-type’ AhCre+ Myc/++ and Myc-deficient AhCre+ Mycfl/fl mice were gavaged with either 200 μl of vehicle or 200 mg/kg nutlin-3a (synthesized at the Roche Research Center, Nutley, NJ, USA) twice a day as previously described.33 On day 4 post Cre induction, mice were given a single application of nutlin and irradiated with 14 Gy and collected 6 h following the irradiation.
Tissue isolation
Tissue isolation was carried out as follows: the proximal 7 cm was fixed overnight in methacarn (methanol, chloroform and acetic acid; 4 : 2 : 1) and then paraffin embedded. The following 3 cm was preserved in RNA later (Sigma, Gillingham, UK). The following 5 cm was bundled using surgical tape and fixed in 4% formaldehyde at 4 °C for 24 h before processing. The remainder was fixed in methacarn.
Assaying apoptosis in vivo
Apoptotic bodies were scored from H&E sections. Twenty-five full crypts were scored from a minimum of three mice of each genotype. Apoptosis was confirmed by immunohistochemical staining against active caspase 3 (1:750, R&D systems, Abbingdon, UK).
Immunohistochemistry
Primary antibodies used for immunohistochemistry: P21 (1 : 500, Santa Cruz, Dallas, TX, USA; M19), P53 (1 : 100 MS-104, PAB240 Neomarkers) and P53 (VectorLabs, Peterborough, UK; CM5), CHK1 pS345 (1 : 100, Cell Signalling, Danvers, MA, USA), ATM pS1981 (1 : 500, ROCKLAND 200-301-500), MDM2 (1 : 200, Lab Vision, Waltham, MA, USA; smp14 ms- 291-p1), γ-H2AX (1 : 300 Upstate), MYC (1 : 500, Santa Cruz, N-262, sc764).
Epithelial extractions
To obtain a population of epithelial cells, an epithelial extraction protocol based on Bjerknes & Cheng48 was performed. In brief, 10 cm of small intestine was flushed with water before being tied and everted over a glass rod. Vibration was then applied, and the intestine placed in 10 mM EDTA in Hanks’ Balanced Salt Solution (HBSS; Gibco, Paisley, UK) at 37 °C for 15 min. The intestine was moved into a fresh tube of 10 mM EDTA/HBSS and incubated for a further 15 min. Epithelial cells were collected by centrifugation (2700 × g, 4 °C, 15 min).
Western blot analysis
Protein was extracted from epithelial extracted samples by standard methods using lysis buffer (20 mM Tris-Hcl pH8.0, 2 mM EDTA (pH8.0), 0.5% (v/v) NP-40) containing protease inhibitors (Complete Mini Protease inhibitor tablets, Roche, Burgess Hill, UK) and phosphatase inhibitors (25 mM sodium β-glycerophosphate, 100 mM sodium fluoride, 20 nM Calyculin A, 10 mM sodium pyrophosphate). Solubilised proteins (20 μg) were separated by standard SDS-PAGE on a 10% polyacrylamide separating gel with 5% stacking gel and subsequently transferred to PVDF membrane (Hybond-P, Amersham Biosciences, Buckinghamshire, UK) by standard methods. Primary antibodies and conditions used to probe blots were rabbit anti-MDM2 (1 : 1000; R&D systems AF1244), mouse anti-P53 (1 : 1000; Cell Signalling Technology 1C12), rabbit anti-P21 (1 : 200; Santa Cruz sc479) and mouse anti-β-actin (1 : 5000; Sigma). Appropriate HRP-conjugated secondary anti-rabbit or anti-mouse antibodies were used (Amersham Biosciences).
Abbreviations
- NMNU:
-
N-methyl-N-nitrosourea
- BET:
-
bromodomain and extra-terminal
- IHC:
-
immunohistochemistry
- Gy:
-
gray
References
Askew DS, Ashmun RA, Simmons BC, Cleveland JL . Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene 1991; 6: 1915–1922.
Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M et al. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992; 69: 119–128.
Hoffman B, Liebermann DA . Apoptotic signaling by c-MYC. Oncogene 2008; 27: 6462–6472.
Hermeking H, Eick D . Mediation of c-Myc-induced apoptosis by p53. Science 1994; 265: 2091–2093.
Pelengaris S, Khan M, Evan GI . Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 2002; 109: 321–334.
Finch A, Prescott J, Shchors K, Hunt A, Soucek L, Dansen TB et al. Bcl-xL gain of function and p19 ARF loss of function cooperate oncogenically with Myc in vivo by distinct mechanisms. Cancer Cell 2006; 10: 113–120.
Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL . Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev 1999; 13: 2658–2669.
Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, Lowe SW . INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes Dev 1999; 13: 2670–2677.
Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA et al. Distinct thresholds govern Myc's biological output in vivo. Cancer Cell 2008; 14: 447–457.
Seoane J, Le HV, Massague J . Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 2002; 419: 729–734.
Mitchell KO, Ricci MS, Miyashita T, Dicker DT, Jin Z, Reed JC et al. Bax is a transcriptional target and mediator of c-myc-induced apoptosis. Cancer Res 2000; 60: 6318–6325.
Barsyte-Lovejoy D, Mao DY, Penn LZ . c-Myc represses the proximal promoters of GADD45a and GADD153 by a post-RNA polymerase II recruitment mechanism. Oncogene 2004; 23: 3481–3486.
Rogulski K, Li Y, Rothermund K, Pu L, Watkins S, Yi F et al. Onzin, a c-Myc-repressed target, promotes survival and transformation by modulating the Akt-Mdm2-p53 pathway. Oncogene 2005; 24: 7524–7541.
Maclean KH, Keller UB, Rodriguez-Galindo C, Nilsson JA, Cleveland JL . c-Myc augments gamma irradiation-induced apoptosis by suppressing Bcl-XL. Mol Cell Biol 2003; 23: 7256–7270.
Cannell IG, Kong YW, Johnston SJ, Chen ML, Collins HM, Dobbyn HC et al. p38 MAPK/MK2-mediated induction of miR-34c following DNA damage prevents Myc-dependent DNA replication. Proc Natl Acad Sci USA 2010; 107: 5375–5380.
Sodir NM, Swigart LB, Karnezis AN, Hanahan D, Evan GI, Soucek L . Endogenous Myc maintains the tumor microenvironment. Genes Dev 2011; 25: 907–916.
Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM et al. Modelling Myc inhibition as a cancer therapy. Nature 2008; 455: 679–683.
Muncan V, Sansom OJ, Tertoolen L, Phesse TJ, Begthel H, Sancho E et al. Rapid loss of intestinal crypts upon conditional deletion of the Wnt/Tcf-4 target gene c-Myc. Mol Cell Biol 2006; 26: 8418–8426.
Sansom OJ, Zabkiewicz J, Bishop SM, Guy J, Bird A, Clarke AR . MBD4 deficiency reduces the apoptotic response to DNA-damaging agents in the murine small intestine. Oncogene 2003; 22: 7130–7136.
Sansom OJ, Clarke AR . P53 null mice: damaging the hypothesis? Mutat Res 2000; 452: 149–162.
Bettess MD, Dubois N, Murphy MJ, Dubey C, Roger C, Robine S et al. c-Myc is required for the formation of intestinal crypts but dispensable for homeostasis of the adult intestinal epithelium. Mol Cell Biol 2005; 25: 7868–7878.
Ireland H, Kemp R, Houghton C, Howard L, Clarke AR, Sansom OJ et al. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: effect of loss of beta-catenin. Gastroenterology 2004; 126: 1236–1246.
Marshman E, Ottewell PD, Potten CS, Watson AJ . Caspase activation during spontaneous and radiation-induced apoptosis in the murine intestine. J Pathol 2001; 195: 285–292.
Sansom OJ, Clarke AR . The ability to engage enterocyte apoptosis does not predict long-term crypt survival in p53 and Msh2 deficient mice. Oncogene 2002; 21: 5934–5939.
Clarke AR, Gledhill S, Hooper ML, Bird CC, Wyllie AH . p53 dependence of early apoptotic and proliferative responses within the mouse intestinal epithelium following gamma-irradiation. Oncogene 1994; 9: 1767–1773.
Peukert K, Staller P, Schneider A, Carmichael G, Hanel F, Eilers M . An alternative pathway for gene regulation by Myc. Embo J 1997; 16: 5672–5686.
Wilkins JA, Sansom OJ . C-Myc is a critical mediator of the phenotypes of Apc loss in the intestine. Cancer Res 2008; 68: 4963–4966.
Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR et al. Myc deletion rescues Apc deficiency in the small intestine. Nature 2007; 446: 676–679.
Pusapati RV, Rounbehler RJ, Hong S, Powers JT, Yan M, Kiguchi K et al. ATM promotes apoptosis and suppresses tumorigenesis in response to Myc. Proc Natl Acad Sci USA 2006; 103: 1446–1451.
Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM . A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 2000; 10: 886–895.
Montes de Oca Luna R, Wagner DS, Lozano G . Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995; 378: 203–206.
Jones SN, Roe AE, Donehower LA, Bradley A . Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995; 378: 206–208.
Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci USA 2006; 103: 1888–1893.
Vassilev LT . MDM2 inhibitors for cancer therapy. Trends Mol Med 2007; 13: 23–31.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303: 844–848.
Wang X, Cunningham M, Zhang X, Tokarz S, Laraway B, Troxell M et al. Phosphorylation regulates c-Myc's oncogenic activity in the mammary gland. Cancer Res 2011; 71: 925–936.
Ashton GH, Morton JP, Myant K, Phesse TJ, Ridgway RA, Marsh V et al. Focal adhesion kinase is required for intestinal regeneration and tumorigenesis downstream of Wnt/c-Myc signaling. Dev Cell 2010; 19: 259–269.
Myant K, Sansom OJ . Wnt/Myc interactions in intestinal cancer: partners in crime. Exp Cell Res 2011; 317: 2725–2731.
Slack A, Chen Z, Tonelli R, Pule M, Hunt L, Pession A et al. The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proc Natl Acad Sci USA 2005; 102: 731–736.
Saxena A, Rorie CJ, Dimitrova D, Daniely Y, Borowiec JA . Nucleolin inhibits Hdm2 by multiple pathways leading to p53 stabilization. Oncogene 2006; 25: 7274–7288.
Cao X, Bennett RL, May WS . c-Myc and caspase-2 are involved in activating Bax during cytotoxic drug-induced apoptosis. J Biol Chem 2008; 283: 14490–14496.
Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci USA 2011; 108: 16669–16674.
Luche H, Weber O, Nageswara Rao T, Blum C, Fehling HJ . Faithful activation of an extra-bright red fluorescent protein in "knock-in" Cre-reporter mice ideally suited for lineage tracing studies. Eur J Immunol 2007; 37: 43–53.
de Alboran IM, O'Hagan RC, Gartner F, Malynn B, Davidson L, Rickert R et al. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity 2001; 14: 45–55.
Deng C, Zhang P, Harper JW, Elledge SJ, Leder P . Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995; 82: 675–684.
Vooijs M, Jonkers J, Berns A . A highly efficient ligand-regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO R 2001; 2: 292–297.
Zhang X, Farrell AS, Daniel CJ, Arnold H, Scanlan C, Laraway BJ et al. Mechanistic insight into Myc stabilization in breast cancer involving aberrant Axin1 expression. Proc Natl Acad Sci USA 2012; 109: 2790–2795.
Bjerknes M, Cheng H . Methods for the isolation of intact epithelium from the mouse intestine. Anat Rec 1981; 199: 565–574.
Acknowledgements
This work was funded by Cancer Research UK, AICR, BBSRC and NH&MRC of Australia (#603127). We thank Dave Gillespie and Lye Mun Tho for pCHK1 antibody and protocol, Colin Nixon for Histology, and Genotyping/Biological Services.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article
Cite this article
Phesse, T., Myant, K., Cole, A. et al. Endogenous c-Myc is essential for p53-induced apoptosis in response to DNA damage in vivo. Cell Death Differ 21, 956–966 (2014). https://doi.org/10.1038/cdd.2014.15
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2014.15
Keywords
This article is cited by
-
Phenolic-rich fraction of green tea attenuates histamine-mediated cardiopulmonary toxicity by inhibiting Cox-2/NF-κB signaling pathway and regulating oxidant/antioxidant balance
Beni-Suef University Journal of Basic and Applied Sciences (2024)
-
Loss of phosphatase CTDNEP1 potentiates aggressive medulloblastoma by triggering MYC amplification and genomic instability
Nature Communications (2023)
-
MicroRNA-874 targets phosphomevalonate kinase and inhibits cancer cell growth via the mevalonate pathway
Scientific Reports (2022)
-
Impact of intravesical instillation of a novel biological response modifier (P-MAPA) on progress of non-muscle invasive bladder cancer treatment in a rat model
Medical Oncology (2022)
-
MYC: a multipurpose oncogene with prognostic and therapeutic implications in blood malignancies
Journal of Hematology & Oncology (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}