
Abstract
Histone deacetylase (HDAC) is an emergent anticancer target, and HR23B is a biomarker for response to HDAC inhibitors. We show here that HR23B has impacts on two documented effects of HDAC inhibitors; HDAC inhibitors cause apoptosis in cells expressing high levels of HR23B, whereas in cells with low level expression, HDAC inhibitor treatment is frequently associated with autophagy. The mechanism responsible involves the interaction of HDAC6 with HR23B, which downregulates HR23B and thereby reduces the level of ubiquitinated substrates targeted to the proteasome, ultimately desensitising cells to apoptosis. Significantly, the ability of HDAC6 to downregulate HR23B occurs independently of its deacetylase activity. An analysis of the HDAC6 interactome identified HSP90 as a key effector of HDAC6 on HR23B levels. Our results define a regulatory mechanism that involves the interplay between HR23B and HDAC6 that influences the biological outcome of HDAC inhibitor treatment.
Similar content being viewed by others


Main
Histone deacetylase is a family of enzymes that control the acetylation of chromatin.1 An increasingly large group of proteins connected with different aspects of normal and tumour cell biology are known to be influenced by acetylation.2, 3, 4 As a consequence, the HDAC family has attracted considerable attention as a therapeutic target.2, 3, 5 Indeed, inhibition of HDAC activity is strongly anti-proliferative on tumour cells in vitro, and delays tumour growth in xenograft models.5, 6, 7 An extensive number of clinical trials with HDAC inhibitors in a variety of malignancies are underway, and two HDAC inhibitors, SAHA/Vorinostat and FK228/Romidepsin, have to date been approved for treating a human malignancy, namely cutaneous T-cell lymphoma (CTCL1, 8, 9, 10). However, identifying other malignancies and disease types that are likely to respond favourably to HDAC inhibitors has been hampered, principally because knowledge of the key pathways through which HDAC inhibitors affect tumour cell growth remains limited.9, 11
In previous studies, we undertook through a genome-wide loss-of-function screen to identify genes that have an impact on the sensitivity of tumour cells to HDAC inhibitors.12 We reasoned that genes identified in this way would not only provide important information on key pathways affected by HDAC inhibitors, but also identify potential biomarkers that inform on the tumour response to HDAC inhibitor-based therapies.11, 12 Through this analysis, we identified HR23B as a protein that influences the response and sensitivity of tumour cells to HDAC inhibitors.12 HR23B functions in at least two pathways; nucleotide excision repair (NER) and protein targeting to the proteasome.13, 14, 15 Further studies suggested that the ability of HR23B to engage in proteasomal shuttling underpins its role as a determinant of HDAC inhibitor sensitivity, and it is consistent with this idea that aberrant proteasome activity occurs in tumour cells treated with HDAC inhibitors.12
The potential utility of HR23B as a predictive biomarker was evaluated in the clinical setting by studying its expression in biopsies taken from a group of patients suffering from CTCL that had been treated with vorinostat,8, 16, 17 where there was a good coincidence between HR23B expression and therapeutic response,16 suggesting that HR23B could provide a useful predictive biomarker for identifying CTCL that responds favourably to HDAC inhibitors. However, treating tumour cells with HDAC inhibitors results in a number of different outcomes, which includes apoptosis, senescence, autophagy and cell cycle arrest.2, 18, 19, 20 Given the variability in cellular response, and the potential utility of HR23B as a response-specific biomarker, we were interested to evaluate whether there was a role for HR23B in the diverse cellular outcomes of HDAC inhibitor treatment.
With this objective in mind, we explored the mechanistic role of HR23B in regulating the biological consequence of HDAC inhibitor treatment. Our results indicate that HR23B has impacts on two documented effects of HDAC inhibitors, namely apoptosis and autophagy. Thus, HDAC inhibitors cause apoptosis in cells expressing high levels of HR23B, whereas in cells with low levels of expression, HDAC inhibitor treatment was frequently associated with autophagy. The mechanism responsible involves the interplay between HR23B and HDAC6, and specifically the ability of HDAC6 to counter-balance the pro-apoptotic effect of HR23B. This is achieved through an interaction between HDAC6 and HR23B, which downregulates HR23B, reducing the level of ubiquitinated substrates targeted to the proteasome and ultimately desensitising cells to apoptosis. Significantly, the ability of HDAC6 to downregulate HR23B occurred independently of its deacetylase activity, and a proteomic analysis of the HDAC6 interactome revealed HSP90 as a key effector in the HDAC6-mediated downregulation of HR23B. These results suggest that the interplay between HR23B and HDAC6 is important in regulating the biological outcome of HDAC inhibitor treatment.
Results
HR23B influences autophagy
We evaluated the effect of HDAC inhibitor treatment on the level of markers for autophagy in different cell types. The HDAC inhibitor SAHA caused LC3 cleavage as well as the appearance of LAMP1 and cathepsin D in diverse tumour cell lines, for example HUT78, MYLA (CTCL), A2058, A375 (melanoma), HCT116 and HCT15 (CRC; Figure 1a and Supplementary Figure 1a-d). Treatment with other HDAC inhibitors, including valproic acid (VPA), resulted in similar effects (Figure 1a and Supplementary Figure 1a-d), and the increase in autophagy markers coincided with the visual appearance of autophagosomes in cells (for example shown for SAHA; Figures 1a). Significantly, when cells were co-treated with the inhibitor of autophagy 3-methyladenine (3-MA);21, 22, 23 and SAHA, an enhanced level of apoptosis was apparent (Figure 1b), suggesting that under these experimental conditions, apoptosis and autophagy are separate outcomes of HDAC inhibitor treatment. These results are consistent with previous reports that document the induction of autophagy by HDAC inhibitors at similar concentrations.18, 20

Autophagy and HR23B in cells treated with HDAC inhibitors. (a)HUT78(i) and MYLA(ii) (CTCL), and HCT116(iii) and HCT15(iv) (CRC) cells were each treated with SAHA at the indicated (μM) concentrations, extracts prepared at 18 h and immunoblotted with anti-HR23B, anti-LC3, anti-LAMP1, anti-cathepsin D or anti-actin antibody as indicated; note that the appearance of the lower form (LC3-II) and increased overall LC3 level is indicative of autophagy. Visualisation of autophagosomes in U2OS cells by immunostaining with anti-LC3 (in red) in SAHA (10 and 20 μM) treated cells is also shown (v). Counter-stain with DAPI was superimposed for each image. (b) U2OS cells were treated with SAHA at the indicated (μM) concentration together with 3-methyladenine (3-MA; 10 mM) for 18 h, when cell extracts were immunoblotted with the indicated antibodies. (c) (i) U2OS cells stably expressing inducible HR23B were either untreated or treated with SAHA as indicated (in μM for 18 h) under non-induced (−) or induced (+) doxycycline treatment conditions (1 μg/ml; induction for 72 h treatment). Cells were harvested and immunoblotted with anti-Flag (for ectopic HR23B), anti-LC3 or actin as indicated. (ii) U2OS stable cells were treated as described in (i) with SAHA (at the indicated concentrations in μM) for 24 h, and the level of apoptosis analysed by immunoblotting for PARP (cleaved (c) and uncleaved) under non-induced (−) or induced (+) doxycycline treatment conditions as indicated. The level of ectopic HR23B and actin is shown. (iii) U2OS cells were treated with HR23B or control (NT) siRNA (50 nM for 72 h) in the absence (−) or presence of SAHA (10 μM) for the indicated times (in h) and the level of PARP (cleaved (c) and uncleaved) measured. The level of HR23B and actin is shown. (d) (i) Autophagosomes in U2OS cells visualised with anti-LC3 (red) stably expressing inducible HR23B (visualised with anti-Flag; green) after treatment with SAHA (10 μM for 18 hr) in the absence (−) or presence (+) of doxycycline. (−) shows the LC3 staining autophagosomes (red) under SAHA treatment, and (+) shows superimposed LC3 (red) and HR23B (green) images. (ii) The levels of HR23B and LC3 were analysed by immunoblotting with anti-Flag (ii), and quantitation of cells with autophagosomes after treatment with SAHA is presented in (iii), in the absence (−) or presence (+) of doxycycline, which was performed in triplicate (error bars indicate S.E.M.). (e) (i) U2OS cells stably expressing HR23B were grown in duplicate in the absence (−) or presence (+) of doxycycline together with SAHA (indicated concentration in μM) and, after 9 days, the number of viable cell colonies assessed by crystal violet staining. The untreated control cells are shown for comparison. (ii) Graphical representation of the data in (i), where values indicate mean ±S.E.M; n=3; *P<0.05 and **P<0.01 Student’s t-test
We noted however that the appearance of autophagocytic markers frequently coincided with decreased levels of HR23B (Figure 1a and Supplementary Figure 1a-d), and surmised that a relationship might exist between HR23B and autophagy. As previous studies established that high levels of HR23B sensitise cells to apoptosis in response to HDAC inhibitor treatment,12, 16 we examined the influence that different levels of HR23B had on cellular outcome. Thus, we altered the level of HR23B, either by expressing high levels of ectopic HR23B protein in stable cell lines or depleting endogenous HR23B with siRNA, and thereafter assessed the impact on autophagy. Reduced levels of LC3 cleavage and enhanced PARP cleavage occurred in cells expressing ectopic HR23B (Figures 1ci), and decreased levels of HR23B resulted in reduced apoptosis (Figure 1biii and Supplementary Figure 1e). Increased levels of autophagy (measured as the appearance of cleaved LC3) were also apparent in HR23B siRNA-treated cells compared with the control treatment (Supplementary Figure 1f and g), and by immunostaining, the autophagosomes apparent in SAHA-treated cells were less evident when ectopic HR23B was expressed (Figure 1d, quantitated in iii). We further evaluated the role of HR23B by measuring its effect in cell colony formation assays, in the presence and absence of SAHA. The expression of ectopic HR23B (in the stable cell line;+Dox) caused a markedly enhanced growth inhibitory effect of the drug compared with uninduced (−Dox) cells, reflected as a reduction in cell colony formation number (Figure 1e; quantitated below). Thus, HR23B has the ability to influence the biological outcome of SAHA treatment.
HR23B influences the level of ubiquitinated proteins and proteasome activity
HR23B binds and shuttles ubiquitinated cargo proteins to the proteasome,13, 14 and proteasome targeting by HR23B is likely to be important for its role as an HDAC inhibitor sensitivity determinant.12, 16 We evaluated this possibility by studying the level of global ubiquitination in HDAC inhibitor-treated cells, and then the impact of HR23B on ubiquitination and directly on proteasome activity. We expressed His-tagged ubiquitin in transfected cells and assessed the level of ubiquitin-conjugated proteins by immunoblotting with anti-His antibodies24 under conditions of drug treatment that causes growth inhibition.12, 16
The global ubiquitination pattern in SAHA-treated (for 20 h) cells was similar to control untreated cells (Figure 2a). An increase in ubiquitination was apparent when the ubiquitin proteasome system (UPS) was directly impaired by treating cells with MG132, and cells treated with both SAHA and MG132 had an even greater level of ubiquitination (Figures 2a and b, quantitation of ubiquitin signal shown below; ii). Upon the expression of ectopic HR23B (+Dox) in the stable cells and treatment with either agent alone or combined, a higher level of global ubiquitination was apparent when compared with the uninduced (−Dox) cells (Figure 2a). Thus, a greater amount of ubiquitinated protein was present in cells under conditions of increased HR23B expression. The increased global ubiquitination pattern evident upon expressing HR23B contrasted with the effect of depleting HR23B with siRNA, where a reduced level of ubiquitinated protein was apparent under single or combined drug treatment conditions (Figure 2b). These results indicate that HR23B has an impact on the amount of ubiquitinated protein present in cells, and further that this coincides with the apoptotic effect of HDAC inhibitors (for example, Figure 1cii).

HR23B has an impact on the ubiquitin proteasome system. (a) (i) U2OS cells stably expressing inducible ectopic Flag-HR23B were transfected with His6-ubiquitin (His-Ub; 2 μg), then treated with doxycycline (+ and −, 1 μg/ml) for 24 h following an additional 20 h treatment with SAHA (5 μM) and/or 4 h treatment with MG132 (20 μM) as indicated. Cell lysates and Ni2+ pull-down eluates were analysed by immunoblotting with anti-His as described. The input HR23B is shown underneath, together with actin levels. (ii) Quantitation of the ubiquitin signal. (b) (i) U2OS cells were transfected with control (NT) or HR23B siRNA (50 nM) for 24 h, then transiently transfected with His6-ubiquitin (His-Ub; 2 μg) for 24 h, following an additional 20 h treatment with SAHA (5 μM) and/or 4 h treatment with MG132 (20 μM) as indicated. Ni2+ pull-down eluates were analysed by immunoblotting with anti-His. The input HR23B level is shown underneath, together with actin levels. (ii) Quantitation of the ubiquitin signal. (c) (i) U2OS cells expressing inducible ectopic Flag-HR23B were transiently transfected with GFPu or GFP (100 ng), then treated with or without doxycycline (+ or −, 1 μg/ml) for 24 h as indicated. Cell lysates were analysed as described. Levels of GFP, HR23B and actin are shown. (ii) Quantitation of the GFP signal. (d) (i) U2OS cells were transfected with control (NT) or HR23B siRNA (50 nM) for 24 h, then transiently transfected with GFP or GFPu (100 ng) for 48 h as indicated. Cell lysates were analysed as described above. Levels of GFP, HR23B and actin are shown. (ii) Quantitation of the GFP signal
In order to directly measure proteasomal activity under different levels of HR23B expression, we studied in transfected cells the level of GFPu, which is a derivative of GFP protein that is targeted to the proteasome by virtue of the Cl1 degron sequence and thus acts as a surrogate and independent measure of proteasomal activity.12, 25 In untreated cells, the GFPu reporter was as expected constitutively degraded, in contrast to the non-proteasome-targeted GFP reporter (Figure 2c), reflecting GFPu targeting to the proteasome.25 In the presence of an increased level of HR23B (expressed in the stable inducible cell line;+Dox), the GFPu level was higher, contrasting with the non-targeted GFP where the level remained unchanged (Figure 2c; quantitation of GFP level shown below). In the converse experiment, depleting HR23B (with siRNA) decreased the level of GFPu, in contrast to the minimal effect on the non-targeted GFP protein that remained unchanged (Figure 2d), implying that reduced levels of HR23B enhance protein degradation. Combined with the previous results (Figures 2a and b), we conclude that global ubiquitination and proteasomal activity is influenced by HR23B, and that the high levels of ubiquitination that occur upon increased HR23B expression most likely impair proteasome function (and conversely low levels of ubiquitination enhance proteasome activity).
Interplay between HR23B and HDAC6
To gather insight on the mechanisms that influence HR23B activity and thereby have an impact on the UPS and cellular outcome of HDAC inhibitors, we considered a role for HDAC6 given it is established to control protein turnover and autophagy.26, 27, 28 We tested whether an interaction between HR23B and HDAC6 was apparent and found in cells that HDAC6 and HR23B interact (Figure 3a); a similar interaction was apparent with ectopically expressed proteins (Figures 4a and b). We then assessed whether HDAC6 was able to influence HR23B protein levels by measuring HR23B in cells stably expressing HDAC6 shRNA, where HDAC6 was virtually undetectable (referred to as KD; Figure 3b). The level of HR23B was markedly higher in the HDAC6-deficient cells compared with the control cells (Figure 3b), contrasting with HR23B RNA that remained similar (Supplementary Figure 1h). Further, an increase in HR23B levels was apparent when A549 cells were transfected with HDAC6 siRNA compared with the control NT siRNA treatment (Figure 3c; compare HDAC6 to HR23B level), and in stable inducible HDAC6-expressing cells, reduced levels of HR23B were apparent upon the expression of HDAC6 (Figure 3d; compare HDAC6 to HR23B level).

Interplay between HDAC6 and HR23B. (a) Extracts prepared from U2OS cells were immunoprecipitated with anti-HR23B anti-HDAC6 or control (C) immunoglobulins, followed by immunoblotting with the indicated antibody. The levels of HR23B and HDAC6 are shown in the input (In), together with the control (C) immunoprecipitation. HR23B and HDAC6 are indicated. (b) Extracts prepared from WT or HDAC6 stable shRNA-expressing (KD) A549 cells were immunoblotted with the indicated antibodies. (c) U2OS cells were transfected with control (NT) or HDAC6 siRNA (50 nM) for 24 h, then transfected with GFP or GFPu for 48 h as indicated. Cell lysates were analysed as described. Levels of GFP, HR23B, HDAC6 and actin are shown. Quantification of the GFP signal is shown underneath. (d) U2OS cells stably expressing inducible ectopic Flag-HDAC6 were treated with doxycycline (+ and −; 1 μg/ml) for 72 h, and transiently transfected with GFPu or GFP (100 ng) for 48 h as indicated. Cell lysates were analysed as described. Quantification of the GFP signal is shown underneath. (e) Either WT or HDAC6 stable shRNA-expressing (KD) A549 cells were treated with SAHA for 24 h as indicated and thereafter the level of sub-G1 (apoptotic) cells measured by flow cytometry (i and ii; n=3, error bars indicate S.E.M.). Extracts were prepared and immunoblotted with antibodies for LAMP1, cathepsin D and LC3 to assess the level of apoptosis and autophagy, in addition to HDAC6, HR23B and actin (iii). (f and g) Lysates from WT and HDAC6 stable shRNA-expressing (KD) cells (f), or WT cells treated with HDAC6, HR23B or NT siRNA for 48 h (g). Cell lysates were analysed by immunoblotting with anti-Ub for endogenous ubiquitination (f and g). Input levels of HDAC6, HR23B and actin are shown underneath. Quantification of the ubiquitin signal is presented in the graph. (h and i) U2OS cells were transfected with control (NT) or HDAC6 siRNA for 48 h, and then immunostained in (h) for LC3 (red) and HDAC6 (green), and in (i) for cleaved PARP (red) and HDAC6 (green). Merged images are shown for each (h and i). Quantification performed by counting 500 cells under NT or HDAC6 siRNA treatment conditions is shown underneath

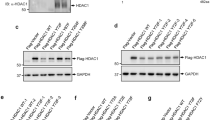
Interaction between HR23B and HDAC6. (a) U2OS cells were transfected with Flag-HDAC6, ΔBUZ, 1–503, ΔN or BUZ (i). Cells were harvested and the HDAC6 mutants were immunoprecipitated with anti-Flag antibody 24 h after transfection and subsequently immunoblotted with anti-Flag or anti-HR23B antibody (ii and iii); note that HR23B is the endogenous protein. Input HDAC6 levels are shown together with actin as a loading control. The C-terminal BUZ region of HDAC6 was required for the interaction with HR23B (note that ΔBUZ was expressed at higher levels compared with WT HDAC6, which could account for the weak co-immunoprecipitated HR23B with ΔBUZ). (b) U2OS cells were co-transfected with Flag-HDAC6 and HR23B (WT) and its different mutants lacking the UbA domains and the UbL domain (i). Cells were harvested and HDAC6 was immunoprecipitated with anti-Flag antibody 24 h after transfection and subsequently immunoblotted with anti-Flag (Flag IB) or anti-Myc antibody (for HR23B). The input level of HDAC6 and HR23B mutants is shown. Only WT HR23B and ΔUbA were co-immunoprecipitated with HDAC6 (ii). Note that the enhanced level of ΔUbA compared with WT HR23B in the immunoprecipitation reflects the increased expression level of the input ΔUbA. (c) Immunoblot (i) showing endogenous HR23B levels upon induction (Dox+treatment) of Flag-HDAC6 in the stable inducible cell line over the indicated time course. (ii) shows endogenous HR23B (red) in the Flag-HDAC6 (green) stable cell line in the same conditions, as indicated at 72 and 96 h post induction (Dox +). DAPI and the merged image is shown. (d and e) WT and HDAC6 stable shRNA-expressing (KD) cells were transfected with HDAC6 (WT) or ΔBUZ (d; 1 and 2 μg), and BUZ (e; 1, 2 and 3 μg) for 72 h as indicated. Cell lysates were analysed by immunoblotting as described with the indicated antibodies. Note that ectopic full-length (WT) HDAC6 is poorly expressed compared to ΔBUZ in transfected cells and is indicated by the arrow (◂) in (d). Endogenous HR23B levels decrease upon expressing WT HDAC6 or BUZ domain, reduced by over 50% by the BUZ domain alone (e; 3 μg BUZ transfection treatment compared with KD treatment)
As the level of HR23B is influenced by HDAC6 (Figures 3b), and as high levels of HR23B sensitise cells to apoptosis (Figure 1c), we reasoned that the absence of HDAC6 might have an impact on cell viability in response to HDAC inhibitors. We evaluated this possibility in the control and HDAC6-deficient (KD) A549 cells. In contrast to the control cells, where a typical low level of apoptosis was apparent and increased apoptosis occurred upon treatment with HDAC inhibitors (about 40-fold), HDAC6-deficient cells had an unusually high basal level of apoptosis (25-fold higher than the control cells). Although a lower relative increase in apoptosis (about two- to three-fold) occurred upon HDAC inhibitor treatment, the absolute level of apoptosis reached was similar to the control cells (Figure 3ei and ii; apoptosis measured as the sub-G1 population by flow cytometry).
We evaluated the level of global ubiquitination in HDAC6 KD cells compared with their WT counterparts, and found that ubiquitination was higher in the KD cells (Figure 3f). The higher level of ubiquitination, and coincident increased level of HR23B in the KD cells (Figure 3f), is compatible with the earlier results describing the higher ubiquitination signal under conditions of increased HR23B expression (Figure 2a). The enhanced sensitivity to apoptosis upon HDAC inhibitor treatment of the KD A549 cells compared with the control cells (Figure 3e) is similarly compatible with the role of HR23B as a positive regulator of HDAC inhibitor-induced apoptosis (Figures 1c). Significantly, enhanced global ubiquitination was also apparent when A549 cells were treated with HDAC6 siRNA compared with the NT siRNA control treatment (Figure 3g). Again, this agrees with the earlier results (Figure 2a), as depleting HDAC6 caused an increase in HR23B that coincided with higher levels of GFPu, but not the untagged GFP version (Figures 3c and g). Most importantly, the increased level of HDAC6 (upon induction in the HDAC6 stable cell line;+Dox) and coincidental decrease in HR23B reflected a reduced level of GFPu, but not GFP (Figure 3d). This was the expected outcome, based on the increase in proteasomal activity and protein turnover (leading to lower levels of GFPu), under conditions of increased HDAC6 and the consequent reduced HR23B levels (Figure 2d).
Autophagy was far more pronounced in the control A549 cells, and the level of autophagy markers reduced in the HDAC6 KD cells compared with the control cells (Figure 3eiii). This effect was also apparent when A549 cells were treated with HDAC6 siRNA, whereupon the number of autophagocytic cells was significantly reduced relative to the control NT siRNA treatment, and contrasted with the number of apoptotic cells that increased under the same HDAC6 siRNA treatment conditions (Figures 3h and i). Overall, these results establish that HDAC6 is a negative regulator of HR23B protein level, which couples HDAC6 activity (via its interplay with HR23B) to the apoptotic and autophagocytic outcome of treatment with HDAC inhibitors.
Regulation of HR23B occurs independently of HDAC6 catalytic activity
We investigated the domains that are required for the interaction between HR23B and HDAC6. Using a series of mutants derived from each protein that were expressed ectopically in cells, the HDAC6 BUZ domain was found to be responsible for the interaction with HR23B because the ΔN and BUZ mutant, both containing the BUZ domain, could interact with HR23B whereas ΔBUZ showed a diminished interaction (Figure 4aii and iii); note that the ΔBUZ–HR23B interaction was not completely abolished most likely because of the high level of ectopic ΔBUZ. Conversely, the UbL domain in HR23B was necessary for the interaction with HDAC6 because the ΔUbA mutant, containing only the UbL domain, could interact with HDAC6 whereas ΔUbL could not (Figure 4bii).
As described earlier (Figure 3d), in cells stably expressing HDAC6, the induction of HDAC6 coincided with reduced levels of HR23B (Figure 4ci); a similar effect on HR23B was evident by immunostaining (Figure 4cii). Moreover, ectopic HDAC6 reduced the level of HR23B in transfected cells (Figure 4d). Significantly, the BUZ domain was required to downregulate HR23B because ΔBUZ failed to alter the level of HR23B (Figure 4d) contrasting with the expression of the BUZ mutant where downregulation of HR23B was evident (Figure 4e). As the BUZ domain is sufficient to affect HR23B, these results equally establish that the catalytic activity of HDAC6 is dispensable for the downregulation of HR23B.
Role of HDAC6 in the apoptosis–autophagy switch
As HDAC6 is a positive regulator of autophagy,28 it appeared possible that HDAC6 may prompt downregulation of HR23B by an autophagocytic process. We therefore assessed the impact of the autophagy inhibitor 3-MA, which failed to affect downregulation of HR23B by HDAC6 (Figure 5a), thus suggesting that HR23B downregulation did not occur through an autophagocytic pathway. Similarly, HDAC6 did not mediate its effects on HR23B via the proteasome, because proteasome inhibitors did not affect the HDAC6-mediated downregulation (Supplementary Figure 1i).

Analysis of HDAC6 interacting proteins. (a) The stable Flag-HDAC6 cell line was treated with (+) or without (−) doxyclycline, and the autophagy inhibitor 3-MA (5 and 10 mM), or SAHA (10 μM) as indicated. (b) Both control cells expressing the vector only (TRE) and stable cells expressing Flag-HDAC6 were induced with doxycyclin (+; 1 μM) for 60 h or left uninduced. Extracts were immunoprecipitated with anti-Flag antibody and subsequently immunoblotted with anti-Flag antibody. Input levels are shown together with actin as loading control, and note under these conditions low level expression of HR23B (a). (c) Co-immunoprecipitation of the Flag-HDAC6 and the empty vector TRE control were stained with a colloidal blue. Dots indicate specific bands in the HDAC6 co-immunoprecipite which were excised and analysed by mass spectrometry. The prominent band around 150 kDa (indicated by the bracket) is the immuno-enriched Flag-HDAC6. Supplementary Table 1 provides further details of the interacting proteins. (d) In silico analysis of HDAC6 interacting proteins found by mass spectrometry are shown for which experimental data have been annotated in STRING.50 The proteins were clustered by the Markov Cluster Algorithm (MCL) and every colour corresponds to one protein cluster. The diameter of the circle represents the betweenness centrality (calculated by the programme Gephi) and is a measure of the importance of the protein in the analysed network. (e) The stable Flag-HDAC6 cell line was treated with (+) or without (−) doxycycline. Extracts were immunoprecipitated with anti-Flag followed by immunoblotting with anti-HSP90. (f) The stable Flag-HDAC6 cell line was treated with (+) or without (−) doxycycline. Extracts were immunoprecipitated with anti-Flag followed by immunoblotting with anti-Ku80/XRCC5 or anti-GRP78/HSPA5. (g) The immunoblot shows the endogenous levels of HR23B upon induction (Dox treatment) of the Flag-HDAC6 stable inducible cell line. Two days after the induction, the HDAC6 specific inhibitor tubastatin and/or (10 μM) the HSP90 specific inhibitor 17-AAG (0.1 or 1 μM) were added and the cells were harvested 2 days later. Acetylated tubulin provides a control for the tubastatin treatment. Extracts were immunoblotted with anti-HSP90, anti-HR23B, anti-tubulin, anti-acetylated tubulin, anti-caspase 3 (cleaved) and anti-actin, as indicated. (h)Model for interplay between HR23B and HDAC6 in regulating the cellular response to HDAC inhibitors. It is envisaged that the level of HR23B and HDAC6 has an impact on the response to HDAC inhibitors, reflected as apoptotic or autophagocytic outcomes of drug treatment (i). The mechanism which underpins HDAC6 and its effect on HR23B does not require catalytic activity but involves the interplay with the HSP90 (via HDAC6 BUZ domain interacting with HR23B UbL domain; (ii). Note that HDAC6 has an established role in activating HSP90 chaperone function through its catalytic (CAT) domain
Thus, in order to explore the mechanism through which HDAC6 downregulates HR23B, we considered the possibility that HDAC6 might mediate its effects by interacting with and perhaps regulating additional proteins. To address this idea, we took a proteomic approach and immunoprecipitated HDAC6 from the induced stable cells (Figure 5b) and analysed the interacting proteome. A variety of proteins were immunoprecipitated with HDAC6 compared with the control treatment (Figure 5c), and subsequently identified by mass spectrometry (presented as protein clusters in Figure 5d and each identified protein detailed in Supplementary Table 1). Some of the proteins had been previously shown to interact with HDAC6, such as β tubulin and its network29 and the molecular chaperones HSP90 and GRP78/HSPA5.30 Other components of the HDAC6 interactome that were newly characterised in this study and occurred repeatedly across different mass spectrometry analyses included the chaperonin TCP1 complex,31 DNA repair proteins like Ku80/XRCC5, PCNA, and MSH2,32 components of the proteasome like PSMD1 (26S proteasome regulatory subunit 1) and metabolic enzymes, like PCK1 (phosphoenolpyruvate carboxykinase 1), suggesting that HDAC6 has the potential to influence a number of different protein networks, and perhaps DNA repair (Figure 5d). These interactions were similarly evident in the inducible HDAC6 stable cells under immunoprecipitation conditions, for example, between HDAC6 and HSP90, GRP78 and Ku80 (Figures 5e and f).
The ability of HDAC6 to bind to interacting partners, like HSP90, suggested the alternative possibility, namely that HDAC6 influences HR23B by targeting this network. Indeed, previous reports have established HDAC6 as an activator of HSP90 chaperone activity.30 Consequently, we evaluated the role of HSP90 in the HDAC6-dependent downregulation of HR23B using the HSP90 inhibitor 17-AAG.33 Notably, HDAC6 failed to downregulate HR23B when HSP90 chaperone activity was inhibited with 17-AAG (Figure 5g). Moreover, the inhibition of apoptosis that occurred upon the expression of HDAC6 (caspase 3 cleavage; Figure 5g) was overcome when HDAC6-induced cells were treated with 17-AAG, which therefore paralled the increase in HR23B levels (Figure 5g). Importantly, although inhibiting HDAC6 catalytic activity (with tubastatin) did not have an impact on the HDAC6-dependent downregulation of HR23B, it did prevent the increase in HR23B upon 17-AAG treatment (Figure 5g), confirming the catalytic independent role of HDAC6 in the downregulation of HR23B, but equally suggesting a role for HDAC6 enzyme activity in the reactivation of HR23B upon treatment with 17-AAG. Thus, HDAC6 is a negative regulator of HDAC inhibitor induced apoptosis through its ability to downregulate HR23B, and further that HDAC6 prompts the downregulation of HR23B through the HSP90 chaperone network.
Discussion
There is increasing evidence that tumour cells evade cell death through autophagy.34, 35, 36 For example, autophagy is initiated by chemotherapy and radiation, where it is believed to represent a tumour cell survival mechanism against stressful agents that induce apoptosis.37, 38 The fact that therapy-induced autophagy provides a survival advantage is supported from studies where pharmacological inhibition of autophagy enhanced the therapeutic effects of drugs in tumour regression models.39, 40, 41 Consequently, autophagy has emerged as a potential tumour cell survival mechanism. Inhibition of HDAC activity has profound anti-proliferative effects, and HDAC inhibitors have different outcomes on cells including induction of apoptosis, cell cycle arrest, senescence, differentiation and, more recently, autophagy.2, 5, 6, 20 However, the mechanisms through which HDAC inhibitors give rise to different biological consequences remain to be determined. Our previous studies identified HR23B as a determinant that enhanced the cellular response to HDAC inhibitors, although it remained unclear whether HR23B could influence the biological outcome of HDAC inhibitor treatment.12 Here, we have established that HR23B has impacts on the outcome of HDAC inhibitor treatment by regulating the switch between apoptosis and autophagy. Within the cell types and systems studied, high levels of HR23B sensitive cells to HDAC inhibitor-induced apoptosis.
HR23B targets the UPS where it shuttles ubiquitinated cargo that are destined for subsequent proteasomal degradation.13, 14 Proteasomal degradation represents one of the most important pathways for regulating proteostasis,42 and one that is well documented as an anticancer mechanism.43 Its integration with other levels of protein turnover, for example, autophagosomal degradation, is also becoming increasingly evident.42, 44, 45, 46, 47 Our results suggest that the ability of HR23B to target the proteasome leads to aberrant proteasome activity that in turn sensitises tumour cells to apoptosis. We identified HDAC6 as a negative regulator of HR23B, defining a mechanism that allows HDAC6 to impinge (via HR23B) on proteasomal activity. As HR23B sensitises tumour cells to apoptosis,12, 16 HDAC6 by virtue of its ability to downregulate HR23B acts to counter-balance the pro-apoptotic activity of HR23B. It is consistent with this idea that HDAC6-deficient cells exhibit higher levels of HR23B, which coincides with increased levels of apoptosis and reduced susceptibility to autophagy. Thus, our results describe a novel level of interplay between HR23B and HDAC6, where the balance between HR23B and HDAC6 is important in dictating the tumour cell effect of HDAC inhibitors.
In exploring the mechanism through which HDAC6 downregulates HR23B, we identified a series of protein networks that potentially could be subject to control by HDAC6. Networks involving tubulins and chaperones were evident, as were others connected with DNA repair and cell metabolism. Indeed, HSP90 was a key effector in the HDAC6-dependent downregulation of HR23B, because the ability of HDAC6 to downregulate HR23B was overcome by treating cells with the HSP90 inhibitor 17-AAG. It is noteworthy that the HSP90 network is responsible for regulating HR23B, because HDAC6 is established to control HSP90 chaperone activity, through a deacetylation event that regulates client protein and chaperone binding.30, 48 We envisage that HDAC6 couples proteasome and chaperone activity, which facilitates its influence on HR23B levels described in this study (Figure 5h).
In conclusion, our studies highlight HR23B as a key regulator of the cellular response to HDAC inhibitors. Given the potential importance of apoptosis and autophagy in cancer and, particularly, the outcome of disease progression, it is clearly possible that HR23B, perhaps in conjunction with HDAC6 and HSP90, provides a biomarker signature with the ability to differentiate between tumours that have differing prognosis (Figure 5h). This information provides a powerful step forward in developing approaches that aim to align the most effective cancer treatment with clinical disease.
Materials and Methods
Cell culture
Cells were cultured at 37 °C in a humidified 5% CO2 incubator in DMEM (Invitrogen, Paisley, Scotland) containing 10% FCS (U2OS, A2058, A375, HCT116, HCT15, WT and shRNA HDAC6 A549 cells), or RPMI containing 20% FCS (HUT78, MYLA). All media contained 1% penicillin/streptomycin (Invitrogen). U2OS-TET-Flag-HR23B and HDAC6 inducible cell lines were grown in DMEM containing 10% tetracycline-negative FCS (PAA Laboratories, GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA), hygromycin (Invitrogen), G418 (Promega, Southampton, UK) and 1% penicillin/streptomycin (Invitrogen). Flag-HR23B and Flag-HDAC6 was induced by the addition of 1 μg/ml doxycycline to culture media. A U2OS-TET-Flag-HDAC6 inducible cell line was created by selecting cells transfected with Flag-HDAC6 inserted in a TET-ON gene expression system (Clontech, Oxford, UK). The control shRNA A549 and HDAC6 KD cells were a kind gift from TP Yao.49 Note that the control A549 cells contain the pSuper plasmid.49
Plasmids
pcDNA-Flag-HDAC6, ΔBUZ, ΔN (439–1215) and 1–503 were a kind gift from TPYao,49 pCMV-GFP-LC3 was from Cell Biolabs Inc. (Cambridge, UK) BUZ was amplified from wild-type pcDNA-Flag-HDAC6 with the following primer combination: forward: 5′-CTACTAGCGGCCGCAATGACCTCAACACGC-3′ and reverse: 5′-TAGTAGGCGGCCGCTTATTTATCATCATC-3′, and then inserted into the pcDNA3.1 vector. pcDNA3.1a-ubiquitin (His6-tagged) was a gift from Dr. Dimitris Xirodimas (CRBM, Montpellier, France). HR23B wild-type and the truncated derivatives ΔUbA and ΔUbL were amplified from wild-type HR23B with the following primer combinations: HR23B wild-type with UbL forward: 5′-GTGGTGGAATTCATGCAGGTCACCCTGAAGACCC-3′ and UbA reverse: 5′-TCTAGACTCGAGATCTTCATCAAAGTTCTGCTGTAG-3′; ΔUbA with UbL forward: 5′-GTGGTGGAATTCATGCAGGTCACCCTGAAGACCC-3′ and UbL reverse: 5′-TCTAGACTCGAGAGACTGACCCGTCACAAGTGC-3′; ΔUbL with UbA forward: 5′-GTGGTGGAATTCATGGCAGTGTCCACACCAGCACCAG-3′ and UbA reverse: 5′-TCTAGACTCGAGATCTTCATCAAAGTTCTGCTGTAG-3′. The PCR products of HR23B wild-type and ΔUbA and ΔUbL mutants were inserted into the pcDNA3.1 (+)/Myc-His vector (Invitrogen) via the restriction sites EcoRI and XhoI using the Liga Fast rapid DNA ligation system (Promega) in order to create myc/his-tagged constructs.
Transfection, siRNA and compound treatment
Cells were transfected with siRNAs against HR23B P (5′-GAUGCAACGAGUGCACUUG-3′), HDAC6 (00349900; Dharmacon, Loughborough, UK) or non-targeting control #2 (Dharmacon, Perbio) as indicated using oligofectamine (Invitrogen) to a final concentration of 50 nM before harvesting. Plasmids were transfected using Genejuice (Merck, Feltham, UK) according to manufacturer instructions. Cells were treated with superoylanilide hydroxamic acid (SAHA), valproic acid, 17-AAG, 3-MA or MG132 (Chemietek, Indianapolis, IN, USA) at the indicated concentrations or conditions and for the indicated times before harvesting.
Purification of proteins modified by ubiquitin
Transfected cells in a 10 cm dish were washed twice with ice-cold PBS and harvested into 1 ml of 8 M urea, 0.1 M Na2HPO4/NaH2PO4, 0.01M Tris-HCl, and pH 8.0. Lysates were sheared by taking through 23-gauge needle to reduce viscosity. Protein concentration was measured with Bradford (Bio-Rad, Hamel Hempstead, UK) and normalised. About 100 μl of lysate was mixed with 4 × SDS loading buffer for input. Then 900 μl of lysate was incubated with 70 μl of a 50% slurry of nickel (Ni2+–NTA)–agarose beads (Qiagen, Manchester, UK), 5 mM imidazole and 10 mM β-mercaptoethanol overnight at 4 °C. The beads were collected by 1 min centrifugation at 8000 r.p.m. and washed with 750 μl of the following buffers with 5 min mixing at room temperature for each washing step: twice with 8 M urea, 0.1M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl, pH 8.0, 10 mM β-mercaptoethanol and 0.1% Triton X-100; three times with 8 M urea, 0.1 M Na2HPO4/NaH2PO4, 0.01M Tris-HCl, pH 6.3, 10 mM β-mercaptoethanol and 0.1% Triton X-100. The bound proteins were then eluted with 100 μl of elution buffer (200mM imidazole, 5% SDS, 150 mM Tris-HCl pH6.8, 30% glycerol, 720 mM β-mercaptoethanol, 0.005% bromophenol blue) for 30 min incubation followed by 10 min spin at 13 000 r.p.m. Samples were heated to 95 °C for 5 min, cooled, and a typical volume of 30 μl loaded on each lane for immunoblot analysis.
Abbreviations
- HDAC:
-
Histone deacetylase
- HSP90:
-
heat-shock protein 90
- CTCL:
-
cutaneous T cell lymphoma
- NER:
-
nucleotide excision repair
- VPA:
-
valproic acid
- 3-MA:
-
3-methyladenine
- GFP:
-
green fluorescent protein
- UPS:
-
ubiquitin proteasome system
- KD:
-
knockdown
- WT:
-
wild-type
- NT:
-
non-targeting
- GRP78:
-
glucose-regulated protein 78
- TCP1:
-
T-complex protein 1
- PCNA:
-
proliferating cell nuclear antigen
- PCK1:
-
phosphoenolpyruvate carboxykinase 1
References
Inche AG, La Thangue NB . Chromatin control and cancer-drug discovery: realizing the promise. Drug Discov Today 2006; 11: 97–109.
Bolden JE, Peart MJ, Johnstone RW . Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 2006; 5: 769–784.
Minucci S, Pelicci PG . Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 2006; 6: 38–51.
Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009; 325: 834–840.
Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK . Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer 2001; 1: 194–202.
Marks PA, Breslow R . Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol 2007; 25: 84–90.
Richon VM, Garcia-Vargas J, Hardwick JS . Development of vorinostat: current applications and future perspectives for cancer therapy. Cancer Lett 2009; 280: 201–210.
Duvic M, Vu J . Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin Investig Drugs 2007; 16: 1111–1120.
Khan O, La Thangue NB . Drug Insight: histone deacetylase inhibitor-based therapies for cutaneous T-cell lymphomas. Nat Clin Pract Oncol 2008; 5: 714–726.
Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R . FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007; 12: 1247–1252.
Stimson L, La Thangue NB . Biomarkers for predicting clinical responses to HDAC inhibitors. Cancer Lett 2009; 280: 177–183.
Fotheringham S, Epping MT, Stimson L, Khan O, Wood V, Pezzella F et al. Genome-wide loss-of-function screen reveals an important role for the proteasome in HDAC inhibitor-induced apoptosis. Cancer Cell 2009; 15: 57–66.
Chen L, Shinde U, Ortolan TG, Madura K . Ubiquitin-associated (UBA) domains in Rad23 bind ubiquitin and promote inhibition of multi-ubiquitin chain assembly. EMBO Rep 2001; 2: 933–938.
Chen L, Madura K . Rad23 promotes the targeting of proteolytic substrates to the proteasome. Mol Cell Biol 2002; 22: 4902–4913.
Schauber C, Chen L, Tongaonkar P, Vega I, Lambertson D, Potts W et al. Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature 1998; 391: 715–718.
Khan O, Fotheringham S, Wood V, Stimson L, Zhang C, Pezzella F et al. HR23B is a biomarker for tumor sensitivity to HDAC inhibitor-based therapy. Proc Natl Acad Sci USA 2010; 107: 6532–6537.
Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007; 109: 31–39.
Shao Y, Gao Z, Marks PA, Jiang X . Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci USA 2004; 101: 18030–18035.
Stimson L, Wood V, Khan O, Fotheringham S, La Thangue NB . HDAC inhibitor-based therapies and haematological malignancy. Ann Oncol 2009; 20: 1293–1302.
Gammoh N, Lam D, Puente C, Ganley I, Marks PA, Jiang X . Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl Acad Sci USA 2012; 109: 6561–6565.
Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y . Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 1998; 23: 33–42.
Seglen PO, Gordon PB . 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA 1982; 79: 1889–1892.
Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P . Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 2000; 275: 992–998.
Camus S, Menendez S, Fernandes K, Kua N, Liu G, Xirodimas DP et al. The p53 isoforms are differentially modified by Mdm2. Cell Cycle 2012; 11: 1646–1655.
Bence NF, Bennett EJ, Kopito RR . Application and analysis of the GFPu family of ubiquitin-proteasome system reporters. Methods Enzymol 2005; 399: 481–490.
Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A et al. HDAC6 is a microtubule-associated deacetylase. Nature 2002; 417: 455–458.
Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007; 447: 859–863.
Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J 2010; 29: 969–980.
Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F . HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol 2008; 18: 291–297.
Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell 2005; 18: 601–607.
Posokhova E, Song H, Belcastro M, Higgins L, Bigley LR, Michaud NA et al. Disruption of the chaperonin containing TCP-1 function affects protein networks essential for rod outer segment morphogenesis and survival. Mol Cell Proteomics 2011; 10: M110 000570.
Lisby M, Barlow JH, Burgess RC, Rothstein R . Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 2004; 118: 699–713.
Trepel J, Mollapour M, Giaccone G, Neckers L . Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 2010; 10: 537–549.
Kroemer G, Marino G, Levine B . Autophagy and the integrated stress response. Mol Cell 2010; 40: 280–293.
Levine B, Kroemer G . Autophagy in the pathogenesis of disease. Cell 2008; 132: 27–42.
Kimmelman AC . The dynamic nature of autophagy in cancer. Genes Dev 2011; 25: 1999–2010.
Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10: 51–64.
Mathew R, Karantza-Wadsworth V, White E . Role of autophagy in cancer. Nat Rev Cancer 2007; 7: 961–967.
Degtyarev M, De Maziere A, Orr C, Lin J, Lee BB, Tien JY et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol 2008; 183: 101–116.
Dang CV . Antimalarial therapy prevents Myc-induced lymphoma. J Clin Invest 2008; 118: 15–17.
Gupta A, Roy S, Lazar AJ, Wang WL, McAuliffe JC, Reynoso D et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc Natl Acad Sci USA 2010; 107: 14333–14338.
Kraft C, Peter M, Hofmann K . Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 2010; 12: 836–841.
Voorhees PM, Orlowski RZ . The proteasome and proteasome inhibitors in cancer therapy. Annu Rev Pharmacol Toxicol 2006; 46: 189–213.
Boyault C, Gilquin B, Zhang Y, Rybin V, Garman E, Meyer-Klaucke W et al. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J 2006; 25: 3357–3366.
Moscat J, Diaz-Meco MT . p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009; 137: 1001–1004.
Korolchuk VI, Menzies FM, Rubinsztein DC . Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 2010; 584: 1393–1398.
Dikic I, Johansen T, Kirkin V . Selective autophagy in cancer development and therapy. Cancer Res 2010; 70: 3431–3434.
Scroggins BT, Robzyk K, Wang D, Marcu MG, Tsutsumi S, Beebe K et al. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol Cell 2007; 25: 151–159.
Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP . The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003; 115: 727–738.
Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J et al. STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 2009; 37 Database issue D412–D416.
Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M . In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 2006; 1: 2856–2860.
David Y, Ternette N, Edelmann MJ, Ziv T, Gayer B, Sertchook R et al. E3 ligases determine ubiquitination site and conjugate type by enforcing specificity on E2 enzymes. J Biol Chem 2011; 286: 44104–44115.
Bastian M, Heymann S, Jacomy M . Gephi: an open source software for exploring and manipulating networks. International AAAI Conference on Weblogs and Social Media 2009.
Acknowledgements
This work was supported by grants from CRUK, MRC, LLR and EU. Heidi Olzscha was supported by an EMBO Long-Term Fellowship, and Maria New by a Gordon Pillar Studentship from LLR. We thank Sarah Atkinson for help in preparing the manuscript.
Author contributions
GL, MN, OK, LS, HO and AC performed the experiments. DK and MM assisted with interpretation. NLT conceived and directed the project, and wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by RA Knight
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
New, M., Olzscha, H., Liu, G. et al. A regulatory circuit that involves HR23B and HDAC6 governs the biological response to HDAC inhibitors. Cell Death Differ 20, 1306–1316 (2013). https://doi.org/10.1038/cdd.2013.47
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2013.47
Keywords
This article is cited by
-
Structure-based prediction of HDAC6 substrates validated by enzymatic assay reveals determinants of promiscuity and detects new potential substrates
Scientific Reports (2022)
-
Posttranslational modification and beyond: interplay between histone deacetylase 6 and heat-shock protein 90
Molecular Medicine (2021)
-
A genome scale RNAi screen identifies GLI1 as a novel gene regulating vorinostat sensitivity
Cell Death & Differentiation (2016)
-
MicroRNA expression profiling predicts clinical outcome of carboplatin/paclitaxel-based therapy in metastatic melanoma treated on the ECOG-ACRIN trial E2603
Clinical Epigenetics (2015)
-
Actin nucleation by WH2 domains at the autophagosome
Nature Communications (2015)
