
Abstract
During mitochondrial apoptosis, pro-apoptotic BH3-only proteins cause the translocation of cytosolic Bcl-2-associated X protein (Bax) to the outer mitochondrial membrane (OMM) where it is activated to release cytochrome c from the mitochondrial intermembrane space, but the mechanism is under dispute. We show that most BH3-only proteins are mitochondrial proteins that are imported into the OMM via a C-terminal tail-anchor domain in isolated yeast mitochondria, independently of binding to anti-apoptotic Bcl-2 proteins. This C-terminal domain acted as a classical mitochondrial targeting signal and was sufficient to direct green fluorescent protein to mitochondria in human cells. When expressed in mouse fibroblasts, these BH3-only proteins localised to mitochondria and were inserted in the OMM. The BH3-only proteins Bcl-2-interacting mediator of cell death (Bim), tBid and p53-upregulated modulator of apoptosis sensitised isolated mitochondria from Bax/Bcl-2 homologous antagonist/killer-deficient fibroblasts to cytochrome c-release by recombinant, extramitochondrial Bax. For Bim, this activity is shown to require the C-terminal-targeting signal and to be independent of binding capacity to and presence of anti-apoptotic Bcl-2 proteins. Bim further enhanced Bax-dependent killing in yeast. A model is proposed where OMM-tail-anchored BH3-only proteins permit passive ‘recruitment’ and catalysis-like activation of extra-mitochondrial Bax. The recognition of C-terminal membrane-insertion of BH3-only proteins will permit the development of a more detailed concept of the initiation of mitochondrial apoptosis.
Similar content being viewed by others


Main
The key step in mitochondrial apoptosis, the release of cytochrome c from the intermembrane space into the cytosol, is controlled by the Bcl-2 family of proteins,1 consisting of the anti-apoptotic group of Bcl-2-like proteins (Bcl-2, B-cell lymphoma-extra large (Bcl-XL), Bcl-2-like protein 2 (Bcl-w), myeloid cell leukemia sequence 1 (Mcl-1), Bcl-2-related protein A1 (A1)), the pro-apoptotic effector group (Bcl-2-associated X protein (Bax) and Bcl-2 homologous antagonist/killer (Bak)) and the pro-apoptotic initiator group, known as BH3-only proteins (Bcl-2-interacting mediator of cell death (Bim), Bid, p53-upregulated modulator of apoptosis (Puma), phorbol-12-myristate-13-acetate-induced protein 1 (Noxa), Bcl-2-interacting killer (Bik), Bcl-2-associated death promoter (Bad), Bcl-2-modifying factor (Bmf) and activator of apoptosis hara-kiri (Hrk)).2
BH3-only proteins act upstream to activate Bax and/or Bak whereas Bcl-2-like proteins interfere with this activation. There is no agreement on how BH3-only proteins activate Bax/Bak. Two models are often quoted.3, 4 The direct activation model is based on the observation that peptides derived from the BH3 domains of BH3-only proteins can activate Bax/Bak and cause the release of cytochrome c from mitochondria. Only peptides representing the BH3 domain of Bid or Bim were active (‘activators’).5 The displacement model derives from data showing that the BH3 domains of different BH3-only proteins have differing affinities for Bcl-2-like proteins, and a combination of BH3 domains may be required to bind to and to neutralise all Bcl-2-like prosurvival proteins.6, 7, 8
Mitochondrial cytochrome c is released upon the functional disintegration of the outer mitochondrial membrane (OMM), achieved through the oligomerisation of Bax or Bak.8 Bak is a tail-anchored protein in the OMM (and the endoplasmic reticulum (ER)). Bax is at least predominantly cytosolic but during apoptosis translocates to mitochondria by an unknown mechanism.
As BH3-only proteins are involved in the activation of mitochondrial Bak and activation/translocation of Bax to mitochondria, their localisation is likely to be of great importance; BH3-only proteins may activate Bax in the cytosol to translocate to mitochondria or may activate Bax when it is already localised at mitochondria. Localisation of proteins to mitochondria usually is not random, but regulated. Proteins may localise to mitochondria because of binding to mitochondrial partners or because they are directed to and ‘imported’ into mitochondria by a specialised machinery. This mitochondrial import is mediated by specific targeting signals within the proteins. Classes of OMM proteins include proteins anchored with a single transmembrane domain either at the N- or C-terminus (the latter called tail-anchored proteins). This domain acts both as a hydrophobic anchor for membrane insertion and as a specific OMM-targeting signal.9
Some studies have reported mitochondrial targeting of the BH3-only proteins Noxa,10 tBid (caspase-8-cleaved, active Bid)11, 12 and BimS (a rarely expressed splice form of Bim).13 This may occur through binding to mitochondrial Bcl-2-like proteins or through mitochondrial import of these proteins in their own right. Structural analyses of BH3-only proteins have varyingly reported transmembrane domains in BH3-only proteins. A recent review, for instance, suggests transmembrane domains only for Bik and Hrk.14 Membrane targeting of, in particular, activator BH3-only proteins is typically not considered.
We tested the possibility that BH3-only proteins are tail-anchored OMM proteins. Five BH3-only proteins, including Bim, tBid and Puma, fulfilled all tested criteria for C-terminally (tail-) anchored OMM proteins. One (Bik) was found to localise to the ER. Importantly, once inserted in the OMM, Bim, tBid and Puma were able to activate extra-mitochondrial Bax. Closer study demonstrated that this activation mechanism functions in the absence of both interaction with Bcl-2-like proteins and active Bax-translocating mechanisms. These results propose a simplified model of mitochondrial apoptosis where Bax is activated directly at mitochondria by BH3-only proteins in the OMM.
Results
The C-termini of BH3-only proteins are OMM-targeting signals
The sorting information of mitochondrial import signals appears to be encoded in a conserved structural feature and not always in the primary amino-acid sequence. Tail-anchored proteins of the OMM are characterised by a single C-terminally located transmembrane domain of moderate hydrophobicity, 10–20 amino-acid residues in length and flanked by positively charged residues. The tail-anchor domain alone is sufficient for mitochondrial targeting and OMM insertion.15 Tail-anchored proteins with slightly different tail-anchor domains are also found at the ER and in peroxisomes.9
Analysis of seven BH3-only protein sequences by various prediction programmes revealed possible transmembrane domains in all C-termini with the exception of Bad. In five of the six predicted sequences, hydrophobic stretches are 12–16 amino acids long and are flanked by positively charged amino-acid residues (Bim, Bid, Bmf, Noxa and Puma). These C-termini define them as potential tail-anchored OMM proteins (Supplementary Figure S1A). Bik carries a typical ER-tail-anchor signal, characterised by an extended transmembrane domain of higher hydrophobicity at the C-terminus and the lack of flanking charged amino-acid residues (Supplementary Figure S1A).
To test for function of these domains, we used an in vitro mitochondrial import assay. This method allows the detection of mitochondrial association and membrane insertion as well as the discrimination of localisation in mitochondrial compartments (outer membrane, intermembrane space, inner membrane and matrix16). Isolated yeast mitochondria were chosen because yeast does not have Bcl-2 family proteins that could bind BH3-only proteins. However, the mitochondrial import machineries are conserved from yeast to humans, and proteins from either species localise correctly in both17.
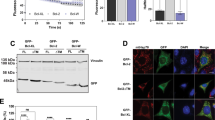
Mouse BH3-only proteins were synthesised by in vitro transcription/translation and subjected to mitochondrial import experiments. When the three splice forms BimEL, BimL and BimS were tested, an association with the re-isolated mitochondria was seen for all three isoforms (Figure 1a, Supplementary Figures S2A, C; the isoforms have the same C-terminus).

In vitro import of BH3-only protein. Import of in vitro-translated BimEL (a), tBid (b), Puma (c), NoxaT103 M (d) or Bad (e) into isolated yeast mitochondria, as described in Materials and Methods. Radiolabelled proteins were incubated with mitochondria isolated from S. cerevisiae cells. Then samples were divided. One part was left untreated (−) and one was treated with 100 μg/ml trypsin (T) for 30 min on ice. Another part was subjected to hypotonic swelling and trypsin treatment (SW/T). The two last parts were subjected to alkaline extraction without or with pre-treatment with trypsin resulting in pellet fractions (P, integral membrane proteins) and supernatant fractions (S, soluble and peripherally attached proteins). For comparison, 10% of the total input of radiolabelled precursors was included (L). Reactions were analysed by SDS-PAGE and autoradiography (f, proteolytic fragments). Molecular weight markers are indicated. Some lanes were digitally removed
The Bim proteins could not be removed from mitochondria by alkaline extraction, indicating integral membrane insertion of the proteins (Figure 1a, Supplementary Figure S2C). The proteins were very likely inserted into the OMM, as they were accessible to protease degradation in intact mitochondria. Tryptic digestion of the import reaction yielded a protected fragment of 4–5 kDa that also remained associated with membranes during alkaline extraction (Figure 1a, Supplementary Figures S2A and C). This fragment very likely corresponds to the C-terminus of Bim that is protected from digestion because of its insertion into the OMM (a peptide of 4.2 kDa is predicted to be generated by tryptic cleavage at residues R163/R164, Supplementary Figure S1A). When Bim proteins were extracted with detergent, protease treatment led to complete digestion of Bim (data not shown). Control experiments with proteins of known submitochondrial localisation are shown in Supplementary Figure S3.
A Bim mutant lacking the predicted tail anchor (BimELΔC; see Supplementary Figure S1A) appeared only loosely attached to rather than inserted into membranes following the import reaction, as the majority of the bound protein was removed by alkaline treatment. Tryptic digestion led to complete degradation of BimELΔC, suggesting that the protected peptide in full-length Bim indeed was the predicted C-terminal membrane anchor (Supplementary Figure S2A).
Green fluorescent protein (GFP) alone did not associate with mitochondria (Supplementary Figure S4A). When fused to the C-terminus of GFP (GFP-Bim-C, Supplementary Figure S1B), the C-terminal peptide of Bim was sufficient to insert the GFP-Bim-C construct into the OMM (Supplementary Figure S2B), in a way very similar to GFP fused to the well-established tail anchor of the mitochondrial outer membrane protein Tom5 (Supplementary Figure S4B). These results are strong evidence that Bim can be inserted as a tail-anchored OMM protein.
Bid is activated by proteolytic cleavage (usually by caspase 8), and the C-terminal fragment (tBid) can induce the Bax/Bak-dependent release of cytochrome c.18, 19 In vitro, tBid inserts into artificial membranes.11 In vitro transcribed full-length Bid became only loosely associated with mitochondria (Supplementary Figure S2D) but in vitro synthesised tBid was C-terminally inserted, as judged by alkaline-resistant association with mitochondrial membranes and retention of a protease-protected fragment in the membrane pellet (Figure 1b).
Full-length Puma (Figure 1c) but not PumaΔC (Supplementary Figure S2E) showed alkaline-resistant membrane insertion, and tryptic digestion left the expected protease resistant, membrane-associated fragments in full-length Puma. The C-terminus of Puma targeted GFP to mitochondria and a similar tryptic fragment was generated as for the full-length Puma protein (Supplementary Figure S2F).
Noxa likewise appeared to be inserted into the OMM (Supplementary Figure S5A). The Noxa-C-terminus further localised GFP to mitochondria where the construct was alkaline-resistant (Supplementary Figure S5B). There is no radioactively labelled methionine in the predicted membrane anchor of Noxa (Supplementary Figure S1A). When a methionine residue was introduced (NoxaT103M (see Supplementary Figure S1A)), a protease-resistant, membrane-integrated fragment appeared in in vitro import experiments (Figure 1d), and a Noxa mutant lacking the C-terminus did not associate with mitochondria (Supplementary Figure S5C). Like Bim, tBid and Puma, Noxa can therefore be inserted into the OMM.
Bmf also appeared to be imported into isolated mitochondria (Supplementary Figure S5D). Little Bad was retained in the mitochondrial fraction, suggestive of association rather than OMM insertion (Figure 1e). Lacking an identifiable OMM anchor and not inserting into mitochondria in vitro, Bad may not be an OMM protein.
The ER-protein Bik20 also showed clear evidence of membrane insertion in the in vitro import experiment (Supplementary Figure S5E), similar to other tail-anchored proteins of the ER that can insert into the mitochondrial outer membrane in vitro21.
C-terminal tail-anchor domains of BH3-only proteins act as mitochondrial targeting signals in intact mammalian cells
We fused the predicted C-terminal tail-anchor signals to the C-terminus of GFP, expressed them in HeLa cells and tested for targeting. GFP alone showed the typical cytosolic and nuclear distribution, whereas the tail-anchor domain of the murine mitochondria fission 1 protein (Fis1) OMM protein targeted GFP to mitochondria as expected (Supplementary Figure S6). The C-terminus of Bim was sufficient to cause at least some mitochondrial localisation of GFP (Figure 2). A portion of GFP-Bim-C localised to ER membranes (Supplementary Figure S7) whereas GFP fused to full-length Bim (Figure 2) or Bim with a mutant BH3 domain was found almost exclusively at mitochondria (Supplementary Figure S6) (for comparison the localisation relative to the ER is for all GFP fusions given in Supplementary Figure S7). Localisation of these proteins was seen around mitochondria, indicative of association with the OMM (see for comparison localisation of GFP targeted to the mitochondrial matrix, Supplementary Figure S6). The C-terminus of Bim therefore appears to be a signal that targets Bim to organelle membranes (predominantly mitochondria), whereas the remainder of Bim (but not an intact BH3 domain) is required for complete mitochondrial localisation.

Subcellular localisation of GFP-fusions as analysed by confocal microscopy. The C-termini of Bim, Puma, Bid, Noxa and Bad (Supplementary Figure S1), or full-length wt BimL were fused to the C-terminus of GFP (GFP-Bim) and the constructs were transiently expressed in HeLa cells. Subcellular localisation of GFP-fusion proteins (green) was analysed by microscopy. Mitochondria were identified by MitoTracker staining (red; Invitrogen, Karlsruhe, Germany). Overlays and details (boxed) are shown. Sizing bar, 10 μm (for overlays) or 1 μm (inset). Over 90% of GFP-positive cells in all experiments showed the pattern described here
The C-terminal domains of Bid, Puma and Noxa showed similar targeting activity whereas the C-terminus of Bik targeted GFP to the ER (as suggested by earlier studies20) (Figure 2, Supplementary Figure S7). The C-terminus of Bad had no clear membrane-targeting activity (Figure 2, Supplementary Figure S7).
We then analysed the localisation of full-length BH3-only proteins upon retroviral transduction of Bax/Bak double-deficient mouse embryonic fibroblasts (MEFs). A substantial part of the three main splice mutants of Bim (BimEL, BimL and BimS) was found in the mitochondrial (heavy membrane) fraction (Figure 3a). Substantial localisation in the mitochondrial fraction was also found for Puma, tBid and Bmf whereas both human and mouse Noxa were found exclusively in the mitochondrial fraction (Figure 3b). Bad was predominantly in the cytosolic fraction (Figure 3b). When tBid was expressed in Bax/Bak-double-deficient MEFs, the picture was similar to the situation for Bim: substantial localisation to both cytosol and mitochondrial fractions was found while the endogenous (intact) Bid was much more strongly localised to the cytosol (Supplementary Figure S8C). A truncation mutant of tBid lacking the C-terminal membrane anchor was detected almost exclusively in the cytosol. Bid lacking the C-terminus was very unstable but the detectable protein localised in a way analogous to full-length Bid (Supplementary Figure S8C).

Localisation of BH3-only proteins in bax−/−/bak−/− DKO MEFs. (a–c) Isolated mitochondria (M) and corresponding cytosolic fractions (c) were analysed by western blotting for the presence of the indicated proteins. After alkaline extraction, membrane-integrated BH3-only proteins stay in the pellet fractions (P) whereas soluble or associated proteins appear in the soluble fractions (S). Hsp60 and cytochrome c (cyt c) served as a positive control for alkaline extraction (S fraction, these soluble matrix proteins are released during the procedure); Caspase 8 served as a control for cytosolic fractions; CoxIV as an integral membrane protein is found in the alkaline resistant mitochondrial fraction (P). (c) Upon FasL treatment Bid is cleaved into tBid, which then becomes inserted into the membrane. This overrepresentation of tBid (as compared with Bid) in the membrane-inserted fraction was quantified using ImageJ. Quantification of Bid or tBid after alkaline extraction (given in %) in the respective fractions (P, S) was calculated from signal intensities of the mitochondrial input fraction (M) set to 100%
When tested for membrane insertion by alkaline extraction of mitochondria isolated from these transgenic cells, the majority of Bim, BH3-mutant Bim, tBid, Puma and Noxa could not be extracted from the mitochondrial membranes, indicating mitochondrial membrane insertion rather than association (Figures 3a and b). The mitochondrial cytosolic distribution of a BH3-domain mutant of Bim, which is incapable of binding to anti-apoptotic Bcl-2 proteins, was unaltered (Figure 3a, Supplementary Figure S8A), indicating that this binding is not relevant for mitochondrial localisation. Deletion of the C-terminal anchor in Bim (Supplementary Figure S1A) however almost abolished mitochondrial localisation (Figure 3a, Supplementary Figure S8A). The addition of ABT-737 (which inhibits binding of BH3-only proteins to Bcl-2, Bcl-XL and Bcl-w22) did not affect localisation of Bim, tBid, Puma or human Noxa (hNoxa) (Supplementary Figures S8A, B). When Bax/Bak double-deficient MEFs overexpressing Bid were treated with FasL, Bid was cleaved and tBid localised in part to the mitochondrial fraction of the cells. As compared with intact Bid, relatively little tBid could be eluted from the membrane fraction using alkaline extraction, again indicating membrane insertion (Figure 3c).
Over expressed Bim was to a substantial part cytosolic (Figure 3a). In HaCaT human keratinocytes and in all three tested human melanoma lines, Bim was almost exclusively found in the mitochondrial fraction (Supplementary Figure S8D). When treated with the drug imiquimod, HaCaT cells undergo Bim-dependent apoptosis.23 Treatment with imiquimod failed to alter subcellular localisation of Bim (Supplementary Figure S8D). When BimS was expressed in the mouse melanoma cell line B16 off an inducible promoter, it initially localised exclusively to mitochondria but was later also in the cytosol (Supplementary Figure S8E). This suggests that Bim is localised primarily at mitochondria but that high levels spill over into the cytosol. Most BH3-only proteins are thus to a substantial part localised at mitochondria.
Mitochondrial BH3-only proteins can activate Bax independently of other Bcl-2 family proteins
As BH3-only proteins thus are OMM proteins, how can they, from this localisation, perform their Bax-activating function, as Bax is a primarily cytosolic protein?24 The simplest model is that BH3-only proteins at mitochondria ‘catalyse’ the activation and insertion of Bax into the OMM. This model would assume that Bim can activate Bax on the mitochondrial membrane. Mitochondrial Bim would then be expected to be able to activate extra-mitochondrial Bax.
We tested this model by expressing Bim in Bax- and Bak-deficient MEFs. Isolated mitochondria from Bax/Bak-deficient MEFs transduced with a control vector failed to release cytochrome c when incubated with recombinant Bax (Figure 4a, right two lanes). However, when mitochondria were used that expressed mouse BimEL, -L or -S, exogenous Bax caused the release of cytochrome c from the mitochondria (Figure 4a). Bim mutants with inactive BH3 domain or without the C-terminal membrane anchor were inactive (Figure 4a). Human BimS (hBimS) showed similar localisation (Figure 3a) and activity (Supplementary Figure S9A).

Bax-induced cytochrome c release from mitochondria expressing Bim, tBid, or Puma. Mitochondrial fractions from bax−/−/bak−/− DKO MEFs were directly tested for their ability to activate Bax to release cytochrome c (cyt c). Recombinant human Bax protein was incubated at the indicated concentrations with mitochondria from MEFs expressing BimEL, BimEL BH3-mutant (BimELΔΔ) and BimEL without the tail anchor (BimELΔC), BimL, BimS and BimS BH3- mutant BimSΔΔ (a), tBid, Puma or human Noxa (hNoxa) (b), or Bad, Bmf and murine Noxa (c). Reactions were split into mitochondria (P) and supernatant (S) by centrifugation, and analysed for cytochrome c-content. Where indicated, 5 μM ABT-737 was added alone or together with Bax for 30 min at 30°C. Western blots are representative of at least two independently performed experiments
tBid- or Puma-expressing mitochondria also released cytochrome c when incubated with Bax (Figure 4b). In contrast, Bad, Bmf and Noxa were inactive (Figure 4c), although we cannot exclude the possibility that the levels of these proteins (expressed from the same promoter as the others) were lower and not high enough for direct activation.
Bim, tBid and Puma are therefore, from their mitochondrial, tail-anchored localisation able to activate extra-mitochondrial Bax. Replacement of the BH3 domain in hBimS with the BH3 domain of Bad (binding Bcl-2, Bcl-w, Bcl-XL but not Mcl-16) abolished its Bax-activating activity (data not shown). The introduction of two additional BH3-domain mutations in this construct restores the binding to all anti-apoptotic Bcl-2-family proteins25 but this failed to restore its Bax-activating activity, although this mutant was integrated into the OMM (Supplementary Figures S9A, S8F). This argues again for a mode of activation of Bax by Bim that is not linked to the binding of Bim to multidomain Bcl-2-family proteins. In the normal situation, such binding may however occur and additionally sensitise cells to other stimuli.
We further expressed Noxa in bax−/−/bak−/− MEFs, isolated mitochondria and tested for Bax-activation in the presence of ABT-737. As Noxa can neutralise Mcl-1 and cause its proteasomal degradation (Supplementary Figure S9B) and ABT-737 can neutralise Bcl-2, Bcl-XL and Bcl-w, this combination blocks all anti-apoptotic Bcl-2-like proteins. However, Bax was not activated on these mitochondria (Figure 4b), arguing against the view that Bax can only be activated by displacement.
Mitochondria were next isolated from bax−/−/bak−/− MEFs and digested with proteinase K (PK) to remove endogenous Bcl-2, Bcl-XL and Mcl-1 (Figure 5b). Exogenous Bax was unable to release cytochrome c from these mitochondria strongly arguing against autoactivation of Bax in the absence of anti-apoptotic Bcl-2 proteins. These mitochondria were then incubated with in vitro translated Bim (see Supplementary Figure S9C). Mitochondria that had in this way imported Bim but not mitochondria loaded with BH3-domain-mutated Bim were able to activate exogenously added Bax to release cytochrome c (Figure 5, lanes + PK). There was no substantial difference between PK-treated and -untreated mitochondria in this assay (Figure 5, compare lanes +/− PK).

Mitochondrial BimEL can activate Bax in the absence of other Bcl-2 family proteins. Mitochondrial fractions from bax−/−/bak−/− DKO MEFs were treated with or without proteinase K (PK), washed and reisolated. Efficiency of PK treatment was analysed by western blot against Bcl-2, Bcl-XL and Mcl-1. Normal levels of cytochrome c and CoxIV indicated intact mitochondria (right panel). In vitro-translated triple HA-tagged BimEL and BimELΔΔ (1 μl) were imported into mitochondria as described in Supplementary Figure S9C. Where indicated, 100 nM recombinant human Bax was added and cytochrome c release was analysed by subsequent SDS-PAGE. P, mitochondrial pellet after Bim import and incubation with or without Bax; S1, supernatant after Bim import (30 min, 30°C); S2, supernatant after Bax import (30 min 30°C); P*, pellet without mitochondria; SN, supernatant after Bim incubation without mitochondria; *cross-reactive band with cytochrome c antibody (probably wheat germ cytochrome c). Western blots are representative of at least two independently performed experiments
These results are strong evidence that activation of extra-mitochondrial Bax by Bim can occur at the OMM in the absence of other detectable Bcl-2 proteins.
We have reported that BimS can sensitise yeast (Saccharomyces cerevisiae) cells to Bax-dependent growth inhibition and mitochondrial hyperpolarisation,13 and the co-expression of Puma and Bax (but not either alone) has been shown to kill yeast cells.26 When Bax was expressed under a tetracycline-dependent promoter, this strongly reduced colony formation, very likely through mitochondrial damage (Supplementary Figure S10). Constitutive expression of BimEL in yeast failed to reduce growth on its own but enhanced the growth-suppressing effect of Bax; this effect was not seen when a Bim BH3-domain-mutant, a mutant lacking the C-terminal anchor domain or GFP was used (Supplementary Figure S10). This suggests the activation of Bax by Bim in the genetic absence of anti-apoptotic Bcl-2-like proteins.
Discussion
We show that BH3-only proteins are at least to a substantial part tail-anchored in the OMM. This mitochondrial localisation is independent of other Bcl-2-proteins and is (in the case of Bim, tBid and Puma) sufficient for the initiation of Bax-dependent release of cytochrome c. No other cytosolic components were required, demonstrating that passive, random diffusion of Bax is sufficient for its mitochondrial translocation and activation. No additional OMM proteins such as Bcl-2-family members were further required. The role of BH3-only proteins in the OMM is therefore likely simply to permit the activation of Bax at the right place.
Peptides comprising the BH3 domain of Bim or Bid have been shown to activate Bax in cell-free systems, although micromolar concentrations of such peptides are required.27 Direct binding by a chemically modified Bim BH3 peptide to Bax has also recently been demonstrated.28, 29 Although in vitro synthesised full-length Bim, Bid and Puma have been coimmunoprecipitated with Bax,30 this has not been achievable in intact cells (only BimS and tBid can be coimmunoprecipitated with Bax but a mutation that disrupts this interaction did not destroy the activity of the BH3-only proteins7). The observed activation of Bax by BH3 peptides, together with the lack of detectable direct interaction between the proteins has engendered the ‘hit-and-run’ model of Bax activation, which proposes a transient interaction of BH3-only proteins with Bax.8
We suggest that a direct, transient interaction between BH3-only proteins and Bax at the mitochondrial outer membrane triggers the mitochondrial insertion of Bax. The mitochondrial transport of Bax then relies on its normal cytosolic diffusion; the role of BH3-only proteins is to facilitate the activation of Bax at the correct location, namely the OMM (Supplementary Figure S11). It is easily conceivable that a transient interaction of membrane-inserted Bim/tBid/Puma with Bax induces the well-known conformational changes in Bax.31 This conformational change may permit the mitochondrial insertion of Bax, and the process is repeated with the next Bax molecule, until enough Bax was inserted to permit oligomerisation; the process would resemble a catalytic activation of Bax through BH3-only proteins. Assuming a transient interaction between BH3-only proteins and Bax and a transient conformational change of Bax before it refolds into its inactive conformation, this model predicts the need for mitochondrial localisation of BH3-only proteins; only Bax that was close to the OMM could insert whereas any Bax in the cytosol might simply refold. This is in accordance with a recent study showing that tBid has to interact with artificial membranes before it can directly interact with Bax (measured by FRET) and cause membrane insertion of Bax, Bax oligomerisation and membrane permeabilisation.11 Although we did not test this directly here, those data in a cell-free system also suggest that the nature of the membrane (that is, mitochondrial versus especially ER membrane) is not important. It is therefore in principle conceivable that Bik at the ER can also activate Bax, but the relevance of this activation would not be clear.
Unlike Bax, Bak is also in its active form at mitochondria where it is inserted into the OMM. For Bak it seems likely that it is kept inactive by binding to other proteins, and the means of its activation may at least in part be different.
BH3-only proteins have been described as largely unstructured when expressed in bacteria.32 However, they may not be unstructured in mammalian mitochondria. Synthetic Bim and Bid BH3 peptides can activate Bax, and this activation involves conformational changes in Bax prior to the insertion into the OMM. BH3 peptides work only at high (micromolar) concentrations. If factors such as conformation of BH3-only proteins and spacing between BH3 domain and mitochondrial membrane in OMM-imported BH3-only proteins were important, these features might be compensated for by high concentrations of the BH3 peptide.
In summary, our data are evidence that Bim, tBid and Puma are mitochondrial proteins that permit Bax activation upon their insertion into the OMM via a tail anchor. We suggest that this occurs through direct interaction with Bax at mitochondria and by catalysing the insertion of Bax into the OMM. Additional functions of BH3-only proteins may still involve the neutralisation of anti-apoptotic Bcl-2-like proteins, perhaps more so in the activation of Bak. However, one key function of Bim, tBid and Puma is probably the recruitment of cytosolic Bax without the requirement of other Bcl-2 family members.
Materials and Methods
Cell lines and, culture conditions and FasL stimulation
MEF deficient for Bax and Bak (bax−/−/bak−/− DKO; immortalised with SV40 large T antigen) were kindly provided by Dr. David Huang, Walter and Eliza Hall Institute (WEHI), Melbourne, Australia. HeLa cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal calf serum (FCS) and antibiotics. For MEF cells, 50 μM of 2-mercaptoethanol was added. B16-BimS cells, HaCaT keratinocytes, and human melanoma cells 1205Lu, WM35 and Sbcl2 were cultured as described earlier.23, 33 All cultures were incubated under standard culture conditions (37°C, 5% CO2). FasL ligand was kindly provided by Dr. Christoph Borner (ZBMZ, Freiburg, Germany). It was obtained by harvesting cell culture media from cells expressing FasL. As a negative control, growth medium was used from cells harboring only the empty expression vector.
Construction of BH3-only expression vectors
The genes coding for the murine BH3-only proteins or corresponding parts as well as the control proteins (the tail-anchor domains of murine Fis1 (amino acid 118–152; GFP-Fis1-C) and yeast Tom5 (amino acid 23–50; GFP-Tom5-C) and the mitochondrial matrix-targeting signal of murine ornithine transcarbamoylase (matrix-GFP)) were amplified by PCR and subcloned into pGEM4 (Promega, Mannheim, Germany) or pcDNA6.2-V5-DEST (Invitrogen) vectors. GFP fusion proteins with tail-anchor domains were generated by overlap extension PCR. Point mutations were generated with the QuickChange II Site-Directed Mutagenesis Kit (Stratagene, Böblingen, Germany). Primer sequences are available on request.
Purification of yeast mitochondria and mitochondrial protein import
Mitochondria were isolated from the Saccharomyces strain D273-10B (ATCC, Manassas, VA, USA, Nr. 24657,) and were obtained by differential centrifugation.34 35S-methionine-labelled precursor proteins were synthesised by in vitro transcription/translation (TNT SP6 Coupled Reticulocyte Lysate, Promega) and incubated with mitochondria in import buffer (600 mM sorbitol, 10 mM MOPS/KOH pH 7.2, 80 mM KCl, 5 mM MgCl2, 2 mM KH2PO4, 5 mM methionine, 3% (w/v) bovine serum albumin, 2.5 mM ATP and 5 mM NADH) for 10 min at 25°C. After treatment of the import reaction with trypsin (100 μg/ml for 30 min on ice, stopped by addition of 1 mg/ml soy bean trypsin inhibitor), mitochondria were re-isolated and analysed by SDS-PAGE and autoradiography. For alkaline extraction, mitochondria were incubated in 100 mM sodium carbonate (30 min on ice), and pellet and supernatant fractions were separated by ultracentrifugation (100 000 × g) at 4°C for 30 min. Import experiments were analysed by tricine-SDS-PAGE35 and autoradiography.
Confocal microscopy
Plasmids were transiently transfected into HeLa cells using Lipofectamine 2000 (Invitrogen). Cells were then seeded onto coverslips for overnight culture, and mitochondria were stained with 20 nM MitoTracker Orange CMTMRos (Molecular Probes, Invitrogen, Karlsruhe, Germany) for 15 min. ER was identified by using the expression plasmid pDsRed2-ER (Clontech, Saint-Germain-en-Laye, France). Cells were imaged with a laser-based spinning disk confocalmicroscope (Andor Technology, Belfast, Northern Ireland), built from an iMIC body (TiLL/Agilent, Gräfelfing, Deutschland), aCSU22 spinning disk (Yokogawa, Amersfoort, The Netherlands), diode-pumped solid-state lasers (488 and 561 nm). Filtered images (Semrock emission filters in a Sutter filter wheel, Rochester, NY, USA) were recorded with a D-977 iXon EMCCD+ camera after twofold magnification (Andor Technology) by using a 100 × objective with a numerical aperture of 1.45 (Olympus, Tokyo, Japan), resulting in a measured pixel size of 0.086 μm at a magnification of 200 × . Stacks were recorded with a coat thickness of 300 μm. Images were processed using the SVI Huygens Professional software and the MBF ImageJ for microscopy collection of plug-ins (http://www.macbiophotonics.ca/imagej).
Retroviral constructs and retroviral infection of MEFs
Genes were subcloned from the pENTR/SD/D-TOPO vector (Invitrogen) into a Gateway compatible retroviral pMIG expression vector. Point mutations were introduced using the Quickchange Lightning Site-Directed Mutagenesis Kit (Strategene, primer sequences available on request).
Retroviral particles were produced in Phoenix-ECO cells by transfection together with packaging vector pCLEco. Bax−/−/bak−/− genes transduced were BimEL (all Bim genes were based on the unspliceable mutant36), BimELΔΔ (BH3-domain deletion of amino acids L150/I153), BimELΔC (last 22 amino acids (tail anchor) are missing), BimL, BimS, BimSΔΔ (deletion of amino-acid residues L64/I6713), Bid, BidΔC or tBidΔC (last 46 amino acids (tail anchor) are missing); tBid, Puma, Bad, Noxa, Bmf, hBimS or hBimSBadY110I/S118G (hBimSBadmut2; amino-acid substitutions in the wild-type Bad-BH3 domain at the same position as in the hBimSBad chimera (mut2) enable the Bad to bind to all Bcl-2 family proteins25). Cells were analysed 48 h after transduction for GFP expression, and if necessary were sorted for GFP expression to achieve a population of >90% GFP-positive cells.
Subcellular fractionation of cell lines
MEFs, HaCaT cells (with and without 50 μg/ml imiquimod, (InvivoGen, Toulouse, France)), B16-BimS cells or human melanoma cell lines were washed once in PBS and resuspended in MB-EDTA buffer (210 mM mannose, 70 mM sucrose, 10 mM HEPES pH 7.4, 1 mM EDTA). Cells were lysed by repeated vigorous resuspending with a 27G syringe.37 Subcellular localisation of Bcl-2 family members was analysed by loading equal volumes of the mitochondria-enriched fraction (after centrifugation at 10 000 × g) and the cytosolic fraction after 45 min ultracentrifugation (4°C at 100 000 × g) on SDS-PAA gels. Caspase-8, Hsp60, tubulin and the CoxIV protein served as marker proteins for cytosolic and mitochondrial fractions. For alkaline extraction, mitochondria (50 μg) were incubated in 400 ìl freshly prepared 100 mM sodium carbonate (pH 11.5). After incubation on ice for 30 min, samples were centrifuged (100 000 × g for 45 min at 4°). Pellet was directly resuspended in Laemmli buffer. Supernatants were precipitated with 12% (v/v) trichloroacetic acid (30 min on ice). Proteins were solubilised in Laemmli buffer, separated by SDS-PAGE and analysed by western blotting.
In vitro translation of BH3-only proteins, import into mitochondria and activation of Bax
In vitro translation was done using the RTS 100 Wheat Germ CECF Kit according to the manufacture instructions (Roche, Mannheim, Germany). PCR was done with the RTS Wheat Germ Linear Template Generation Set (Roche). Mouse mitochondria (30 μg) were treated with or without 25 μg/ml PK (PK, Sigma, Munich, Germany) for 20 min on ice in MB-EDTA buffer and reactions were stopped with 4 mM phenylmethylsulfonylfluoride (PMSF) (10 min, ice). Mitochondria were re-isolated and used for Bim import. For Bim import and activation of purified human Bax, 1 μl of in vitro translated protein was added to these mitochondria.
Cytochrome c-release assay
Directly after the isolation of mitochondria from bax−/−/bak−/− DKO MEFs, aliquots of 30 μg mitochondria were pelleted by centrifugation (10 000 × g, 10 min, 4°C). In the next step, KCl buffer (10 mM HEPES pH 7.4, 125 mM KCl, 4 mM MgCl2, 5 mM KH2PO4, 0.5 mM EGTA) containing different concentrations of purified Bax protein were added (0–500 nM; kindly provided by Dr. Jean-Claude Martinou, University of Geneva, Geneva, Switzerland and Dr. Kornelius Zeth, Tuebingen, Germany), and mitochondria were incubated for 30 min at 30°C in a volume of 50 μl. Upon centrifugation (10 000 × g, 10 min, 4°C), pellet was washed with 150 μl KCl buffer followed by addition of equal volumes of Laemmli buffer. Cytochrome c and human Bax protein were detected by western blot.
Western blotting
Protein concentrations were estimated by the method of Bradford. The following antibodies were used: against Bim, Puma, Bad, Caspase-8, Bcl-2, Bcl-XL, Bax, Hsp60 (Cell Signaling, Beverley, MA, USA), Noxa (Abcam, Cambridge, UK; Alexis, San Diego, CA, USA), Bid/tBid (8C3 & 2D1 clone kindly provided by Dr. Thomas Kaufmann (Bern) and Dr. David Huang, WEHI), cytochrome c (BD/Pharmingen, Heidelberg, Germany), tubulin (Sigma), Bax (clone 6A7, Upstate Biotechnology, Waltham, MA, USA and Cell Signaling), cytochrome c oxidase subunit IV (CoxIV, MoBiTec, Göttingen, Germany) and Mcl-1 (Rockland, Gilbertsville, PA, USA). Secondary antibodies weres HRPO-coupled anti-mouse (Dianova, Hamburg, Germany) or rabbit (Sigma) IgG). An enhanced chemiluminescence system was used (GE Healthcare, Munich, Germany).
Analysis of cell death in yeast
Standard genetic techniques were used for growth and manipulation of yeast strains.38 Mouse Bax was cloned into the tet-off plasmid pCM189.39 The mouse Bim gene was inserted into the constitutive-expression vector p415-ADH.40 Yeast cells were grown in selective S.D.-medium (S.D.-Ura/-Leu) containing 2% glucose and 1 μg/ml tetracycline. Cells were grown to log phase in synthetic medium lacking uracil and leucin (S.D.-Ura/-Leu), washed three times and diluted in distilled water to an OD600 of 0.5. Cells were diluted in 10-fold steps and spotted on S.D. plates containing 2% glycerol with and without tetracycline (1 μg/ml). Plates were incubated for 4 days at 30° C and imaged with a CCD camera.
Abbreviations
- Bax:
-
Bcl-2-associated X protein
- Bak:
-
Bcl-2 homologous antagonist/killer
- Bcl-XL:
-
B-cell lymphoma-extra large
- Bcl-2:
-
B-cell lymphoma 2
- Bcl-w:
-
Bcl-2-like protein 2
- Mcl-1:
-
myeloid cell leukemia sequence 1
- A1:
-
Bcl-2-related protein A1
- Bim:
-
Bcl-2-interacting mediator of cell death
- Bid:
-
BH3-interacting domain death agonist
- PUMA:
-
p53 upregulated modulator of apoptosis
- Noxa:
-
phorbol-12-myristate-13-acetate-induced protein 1
- Bad:
-
Bcl-2-associated death promoter
- Bmf:
-
Bcl-2-modifying factor
- Bik:
-
Bcl-2-interacting killer
- Hrk:
-
activator of apoptosis hara-kiri
- GFP:
-
green fluorescent protein
- Fis1:
-
mitochondria fission 1 protein
- Hsp60:
-
heat shock protein 60
- CoxIV:
-
cytochrome c oxidase IV subunit
- DMEM:
-
Dulbecco's Modified Eagle's Medium
- DMSO:
-
dimethyl sulfoxide
- FCS:
-
fetal calf serum
- PMSF:
-
phenylmethylsulfonylfluoride
References
Hengartner MO . The biochemistry of apoptosis. Nature 2000; 407: 770–776.
Youle RJ, Strasser A . The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9: 47–59.
Chipuk JE, Green DR . How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? TrendsCell Biol 2008; 18: 157–164.
Willis SN, Adams JM . Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol 2005; 17: 617–625.
Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ . Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002; 2: 183–192.
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 2005; 17: 393–403.
Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007; 315: 856–859.
Dewson G, Kluck RM . Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci 2009; 122 (Pt 16): 2801–2808.
Borgese N, Brambillasca S, Colombo S . How tails guide tail-anchored proteins to their destinations. Curr Opin Cell Biol 2007; 19: 368–375.
Woo HN, Seo YW, Moon AR, Jeong SY, Jeong SY, Choi EK et al. Effects of the BH3-only protein human Noxa on mitochondrial dynamics. FEBS Lett 2009; 583: 2349–2354.
Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B et al. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 2008; 135: 1074–1084.
Billen LP, Kokoski CL, Lovell JF, Leber B, Andrews DW . Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol 2008; 6: e147.
Weber A, Paschen SA, Heger K, Wilfling F, Frankenberg T, Bauerschmitt H et al. BimS-induced apoptosis requires mitochondrial localization but not interaction with anti-apoptotic Bcl-2 proteins. J Cell Biol 2007; 177: 625–636.
Hardwick JM, Youle RJ . SnapShot: BCL-2 proteins. Cell 2009; 138: 404 404 e1.
Horie C, Suzuki H, Sakaguchi M, Mihara K . Characterization of signal that directs C-tail-anchored proteins to mammalian mitochondrial outer membrane. Mol Biol Cell 2002; 13: 1615–1625.
Mokranjac D, Neupert W . Protein import into isolated mitochondria. Methods Mol Biol 2007; 372: 277–286.
Dolezal P, Likic V, Tachezy J, Lithgow T . Evolution of the molecular machines for protein import into mitochondria. Science 2006; 313: 314–318.
Li H, Zhu H, Xu CJ, Yuan J . Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998; 94: 491–501.
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X . Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998; 94: 481–490.
Germain M, Mathai JP, Shore GC . BH-3-only BIK functions at the endoplasmic reticulum to stimulate cytochrome c release from mitochondria. J Biol Chem 2002; 277: 18053–18060.
Schuldiner M, Metz J, Schmid V, Denic V, Rakwalska M, Schmitt HD et al. The GET complex mediates insertion of tail-anchored proteins into the ER membrane. Cell 2008; 134: 634–645.
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435: 677–681.
Weber A, Kirejczyk Z, Potthoff S, Ploner C, Hacker G . Endogenous Noxa determines the strong proapoptotic synergism of the BH3-mimetic ABT-737 with chemotherapeutic agents in human melanoma cells. Transl Oncol 2009; 2: 73–83.
Fletcher JI, Meusburger S, Hawkins CJ, Riglar DT, Lee EF, Fairlie WD et al. Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc Natl Acad Sci USA 2008; 105: 18081–18087.
Day CL, Chen L, Richardson SJ, Harrison PJ, Huang DC, Hinds MG . Solution structure of prosurvival Mcl-1 and characterization of its binding by proapoptotic BH3-only ligands. J Biol Chem 2005; 280: 4738–4744.
Gallenne T, Gautier F, Oliver L, Hervouet E, Noel B, Hickman JA et al. Bax activation by the BH3-only protein Puma promotes cell dependence on antiapoptotic Bcl-2 family members. J Cell Biol 2009; 185: 279–290.
Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell 2005; 17: 525–535.
Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG et al. BAX activation is initiated at a novel interaction site. Nature 2008; 455: 1076–1081.
Gavathiotis E, Reyna DE, Davis ML, Bird GH, Walensky LD . BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol Cell 2010; 40: 481–492.
Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP et al. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell 2009; 36: 487–499.
Hsu YT, Youle RJ . Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem 1997; 272: 13829–13834.
Hinds MG, Smits C, Fredericks-Short R, Risk JM, Bailey M, Huang DC et al. Bim, Bad and Bmf: intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ 2007; 14: 128–136.
Lohmann C, Muschaweckh A, Kirschnek S, Jennen L, Wagner H, Hacker G . Induction of tumor cell apoptosis or necrosis by conditional expression of cell death proteins: analysis of cell death pathways and in vitro immune stimulatory potential. J Immunol 2009; 182: 4538–4546.
Daum G, Bohni PC, Schatz G . Import of proteins into mitochondria. Cytochrome b2 and cytochrome c peroxidase are located in the intermembrane space of yeast mitochondria. J Biol Chem 1982; 257: 13028–13033.
Schagger H . Tricine-SDS-PAGE. Nat Protoc 2006; 1: 16–22.
Shinjyo T, Kuribara R, Inukai T, Hosoi H, Kinoshita T, Miyajima A et al. Downregulation of Bim, a proapoptotic relative of Bcl-2, is a pivotal step in cytokine-initiated survival signaling in murine hematopoietic progenitors. Mol Cell Biol 2001; 21: 854–864.
Eskes R, Desagher S, Antonsson B, Martinou JC . Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol 2000; 20: 929–935.
Sherman F, Fink GR, Hicks J . Methods in Yeast Genetics: A Laboratory Course. Cold Spring Harbor Laboratory Press: New York, 1986.
Gari E, Piedrafita L, Aldea M, Herrero E . A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast 1997; 13: 837–848.
Mumberg D, Muller R, Funk M . Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 1995; 156: 119–122.
Acknowledgements
We thank Heike Bauerschmitt (Ludwig-Maximilians-Universität München) and Patrick Neubert for experimental support. This work was supported by grants from the Deutsche Forschungsgemeinschaft DFG (HA 2128/10, SP and GH), the Deutsche Krebshilfe, Dr. Mildred Scheel-Stiftung (GH) and a predoctoral fellowship from the Boehringer Ingelheim Fonds (FW).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by C Borner
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Wilfling, F., Weber, A., Potthoff, S. et al. BH3-only proteins are tail-anchored in the outer mitochondrial membrane and can initiate the activation of Bax. Cell Death Differ 19, 1328–1336 (2012). https://doi.org/10.1038/cdd.2012.9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2012.9
Keywords
This article is cited by
-
Mitochondria supply sub-lethal signals for cytokine secretion and DNA-damage in H. pylori infection
Cell Death & Differentiation (2022)
-
Pyrroloquinoline quinone (PQQ) protects mitochondrial function of HEI-OC1 cells under premature senescence
npj Aging (2022)
-
Chlamydia trachomatis inhibits apoptosis in infected cells by targeting the pro-apoptotic proteins Bax and Bak
Cell Death & Differentiation (2022)
-
Neuronal cell life, death, and axonal degeneration as regulated by the BCL-2 family proteins
Cell Death & Differentiation (2021)
-
Dynein light chain binding determines complex formation and posttranslational stability of the Bcl-2 family members Bmf and Bim
Cell Death & Differentiation (2020)
