
Abstract
Myc oncoproteins are commonly upregulated in human cancers of different organ origins, stabilized by Aurora A, degraded through ubiquitin–proteasome pathway-mediated proteolysis, and exert oncogenic effects by modulating gene and protein expression. Histone deacetylases are emerging as targets for cancer therapy. Here we demonstrated that the class III histone deacetylase SIRT2 was upregulated by N-Myc in neuroblastoma cells and by c-Myc in pancreatic cancer cells, and that SIRT2 enhanced N-Myc and c-Myc protein stability and promoted cancer cell proliferation. Affymetrix gene array studies revealed that the gene most significantly repressed by SIRT2 was the ubiquitin–protein ligase NEDD4. Consistent with this finding, SIRT2 repressed NEDD4 gene expression by directly binding to the NEDD4 gene core promoter and deacetylating histone H4 lysine 16. Importantly, NEDD4 directly bound to Myc oncoproteins and targeted Myc oncoproteins for ubiquitination and degradation, and small-molecule SIRT2 inhibitors reactivated NEDD4 gene expression, reduced N-Myc and c-Myc protein expression, and suppressed neuroblastoma and pancreatic cancer cell proliferation. Additionally, SIRT2 upregulated and small-molecule SIRT2 inhibitors decreased Aurora A expression. Our data reveal a novel pathway critical for Myc oncoprotein stability, and provide important evidences for potential application of SIRT2 inhibitors for the prevention and therapy of Myc-induced malignancies.
Similar content being viewed by others


Main
The Myc family of oncoproteins are commonly upregulated in human cancer. MYCN oncogene amplification and consequent N-Myc oncoprotein overexpression occur in 20–25% of neuroblastoma and correlate with a poor patient outcome.1, 2, 3 MYC oncogene amplification occurs in 54% of human pancreatic cancer cell lines4 and 33% of human primary pancreatic tumors,5 and significant c-Myc oncoprotein overexpression is seen in ∼50% of human primary pancreatic tumors.6
Stabilization and degradation of Myc oncoproteins are controlled by ordered phosphorylation at serine 62 (S62) and threonine 58 (T58) and consequent ubiquitin–proteasome pathway-mediated proteolysis.7, 8, 9 Aurora A interacts with both N-Myc and ubiquitin, and blocks ubiquitin-regulated N-Myc protein degradation.8 Myc oncoproteins induce malignant transformation by binding to cognate DNA sequences and consequently modulating gene transcription10, 11, 12, 13 as well as by enhancing ribosome biogenesis and consequently upregulating protein expression,14, 15 leading to cell proliferation.
Recruitment of histone deacetylase (HDACs) to gene promoters induces histone hypoacetylation and transcriptional repression, particularly of tumor suppressor genes.16 In a comprehensive panel of normal cells, cancer cell lines, normal tissues, and primary tumors, global loss of monoacetylation of histone H4 at lysine 16 (H4K16) is seen only in cancer cells and is associated with early stages of tumorigenesis.17
One of the HDACs that cause H4K16 deacetylation is the class III HDAC SIRT2, which shows a strong preference for acetylated H4K16.18 Mouse embryonic fibroblasts deficient for SIRT2 show higher levels of H4K16 acetylation in mitosis. The enzymatic conversion of acetylated H4K16 to its deacetylated form may be pivotal to the formation of condensed chromatin.19
In the current study, we demonstrate that the Myc oncoproteins N-Myc and c-Myc upregulate SIRT2 expression in neuroblastoma and pancreatic cancer cells. In a positive feedback loop, SIRT2 represses gene transcription of the E3 ubiquitin–protein ligase NEDD4, leading to reduced N-Myc and c-Myc protein ubiquitination and degradation. Additionally, SIRT2 upregulates and small molecule SIRT2 inhibitors decrease Aurora A expression.
Results
Upregulation of SIRT2 by Myc oncoproteins promotes neuroblastoma and pancreatic cancer cell proliferation
Myc oncoproteins not only modulate gene expression by directly binding to target gene promoters,11 but also modulate protein expression by enhancing ribosome biogenesis and mRNA translation to protein.14, 15 As H4K16 deacetylation is a common hallmark of cancer17 and SIRT2 causes H4K16 deacetylation,18 we examined whether Myc modulated SIRT2 gene and protein expression. As shown in Figures 1a and b, transfection of MYCN-amplified BE(2)-C human neuroblastoma cells with two different N-Myc siRNAs (N-Myc siRNA-1 and N-Myc siRNA-2) significantly reduced N-Myc mRNA and protein expression, and transfection of c-Myc overexpressing MiaPaca-2 human pancreatic cancer cells with c-Myc siRNA-1 or c-Myc siRNA-2 significantly reduced c-Myc mRNA and protein expression. While showing no effect on SIRT2 mRNA expression, N-Myc siRNA-1 and N-Myc siRNA-2 reduced SIRT2 protein expression in BE(2)-C cells, and c-Myc siRNA-1 and c-Myc siRNA-2 reduced SIRT2 protein expression in MiaPaca-2 cells (Figure 1b). Consistently, N-Myc siRNAs significantly reduced the expression of SIRT2 protein, but not SIRT2 mRNA, in MYCN-amplified CHP134 human neuroblastoma cells (Supplementary Figures S1A and B). These data demonstrate that N-Myc and c-Myc upregulate SIRT2 protein expression through a post-transcriptional mechanism.

Upregulation of SIRT2 expression promotes neuroblastoma cell proliferation. (a and b) BE(2)-C and MiaPaca-2 cells were transfected with scrambled control siRNA, N-Myc siRNA-1, N-Myc siRNA-2, c-Myc siRNA-1, c-Myc siRNA-2, SIRT2 siRNA-1 or SIRT2 siRNA-2 for 48 h, followed by RNA and protein extraction, real-time RT-PCR (a) and immunoblot (b) analyses of N-Myc, c-Myc, and SIRT2 mRNA and protein expression. (c-e) BE(2)-C and MiaPaca-2 cells were transfected with scrambled control siRNA, N-Myc siRNA-1, N-Myc siRNA-2, c-Myc siRNA-1, c-Myc siRNA-2, SIRT2 siRNA-1 or SIRT2 siRNA-2 (c), or treated with vehicle control, the SIRT2-selective inhibitor AC-93253 (d) or the SIRT1/SIRT2 inhibitor Salermide (e). Seventy-two hours later, relative cell numbers were examined by Alamar blue assays, and expressed as percentage change in cell numbers. Error bars represented S.E. ***P<0.001
We next examined whether upregulation of SIRT2 contributed to a Myc-induced cancer phenotype. Alamar blue assays revealed that N-Myc siRNA-1, N-Myc siRNA-2, c-Myc siRNA-1, c-Myc siRNA-2, SIRT2 siRNA-1 or SIRT2 siRNA-2 all significantly reduced the numbers of viable BE(2)-C, MiaPaca-2 (Figure 1c) and CHP134 cells (Supplementary Figure S1C) in 3 days. Cell cycle analyses revealed that SIRT2 siRNAs consistently reduced the percentage of cells at S phase, but did not consistently increase the percentage of cells at pre-G1 phase (Supplementary Figure S2, Supplementary Tables S1 and S2), indicating that repression of SIRT2 decreased cell proliferation but did not result in cell death. To exclude potential off-target effects of the siRNAs, we treated BE(2)-C and MiaPaca-2 cells with AC-93253, a small molecule SIRT2-selective deacetylation inhibitor,20 or the SIRT1/SIRT2 inhibitor Salermide, which inhibits SIRT2 but not SIRT1 at 25–50 μM.21 As shown in Figures 1d and e, treatment with AC-93253 or 25–50 μM Salermide induced a dose-dependent growth inhibition in BE(2)-C and MiaPaca-2 cells.
SIRT2 stabilizes Myc oncoproteins
Surprisingly, our immunoblot analyses showed that SIRT2 siRNAs reduced N-Myc protein expression in BE(2)-C (Figure 1b) and CHP134 (Supplementary Figure S3A) neuroblastoma cells, and reduced c-Myc protein expression in MiaPaca-2 pancreatic cancer cells (Figure 1b) without affecting N-Myc and c-Myc mRNA expression (Figure 1a and Supplementary Figure S1A). To exclude potential off-target effects of the siRNAs, we treated BE(2)-C and MiaPaca-2 cells with the SIRT2-selective inhibitor AC-93253 at 0.6 μM20 or the SIRT1/SIRT2 inhibitor Salermide at 50 μM, at which Salermide inhibits SIRT2 but not SIRT1.21 Real-time RT-PCR and immunoblot analyses showed that treatment with 0.6 μM AC-93253 or 50 μM Salermide considerably reduced N-Myc and c-Myc protein expression, even though 0.6 μM AC-93253 and 50 μM Salermide increased c-Myc mRNA expression (Figures 2a and b).

SIRT2 upregulates N-Myc and c-Myc protein expression by blocking their degradation. (a and b) BE(2)-C and MiaPaca-2 cells were treated with control, the SIRT2-selective inhibitor AC-93253 at 0.6 μM or the SIRT1/SIRT2 inhibitor Salermide at 50 μM, followed by real-time RT-PCR (a) and immunoblot (b) analyses of N-Myc and c-Myc mRNA and protein expression. Error bars represented standard error. **P<0.01 and P<0.001. (c) BE(2)-C and MiaPaca-2 cells were transfected with scrambled control siRNA, SIRT2 siRNA-1 or SIRT2 siRNA-2 for 48 h, followed by treatment with the proteasome inhibitor MG-132 (10 μM) for 3 h. N-Myc and c-Myc protein expression was analyzed by immunoblot. (d) BE(2)-C and MiaPaca-2 cells were transfected with scrambled control siRNA or SIRT2 siRNA-1 for 30 h, and treated with 50 μM cycloheximide (CHX) for the last 0, 15, 30, 45, or 60 min in BE(2)-C cells and for the last 0, 10, 20, 30, or 45 min in MiaPaca-2 cells. Protein was extracted from the cells and subjected to immunoblot analysis of N-Myc and c-Myc. N-Myc and c-Myc protein levels were normalized by actin, the ratio of N-Myc protein/actin protein as well as the ratio of c-Myc protein/actin protein were artificially set as 1.0 for samples untreated with CHX, and half-life (T1/2) of N-Myc and c-Myc proteins was obtained from the line chart
As Myc oncoproteins are degraded through ubiquitin–proteasome pathway-mediated proteolysis, we treated BE(2)-C and MiaPaca-2 cells with the proteasome inhibitor MG-132 after siRNA transfection. We have previously shown that MG-132 does not increase Myc protein expression in cells transfected with Myc siRNAs, which ablates Myc mRNA.22 As shown in Figure 2c, MG-132 significantly upregulated N-Myc and c-Myc protein expression in BE(2)-C and MiaPaca-2 cells transfected with SIRT2 siRNA-1 or SIRT2 siRNA-2 for 48 h. We next treated the cells with 50 μM cycloheximide at different time points after transfection with control siRNA or SIRT2 siRNA-1 for only 30 h, when the effect of SIRT2 siRNA-1 on Myc protein expression was minimal. Immunoblot analysis showed that N-Myc protein half-life was reduced from ∼50 min in BE(2)-C cells transfected with control siRNA to ∼32 min in BE(2)-C cells transfected with SIRT2 siRNA-1 (Figure 2d). Consistently, immunoblot analysis demonstrated that c-Myc protein half-life was reduced from ∼35 min in MiaPaca-2 cells transfected with control siRNA to ∼20 min in MiaPaca-2 cells transfected with SIRT2 siRNA-1 (Figure 2d). Taken together, these data suggest that SIRT2 reduces ubiquitin–proteasome pathway-mediated Myc protein degradation and consequently stabilizes Myc oncoproteins.
SIRT2 stabilizes Myc proteins without modulating ERK protein phosphorylation and GSK3 protein expression
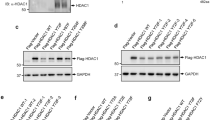
Myc oncoproteins are well known to be stabilized when phosphorylated at S62 by phosphorylated extracellular signal-regulated protein kinase (ERK), and destabilized when phosphorylated at T58due to glycogen synthase kinase 3 (GSK3).7, 8, 23 We therefore examined whether SIRT2 increased Myc protein stability by modulating GSK3 protein expression, ERK protein phosphorylation, and Myc protein phosphorylation. As shown in Figure 3a and Supplementary Figure S3A, transfection of BE(2)-C, MiaPaca-2, and CHP134 cells with SIRT2 siRNA-1 or SIRT2 siRNA-2 reduced S62-phosphorylated, T58-phosphorylated, and total N-Myc and c-Myc protein. Moreover, SIRT2 siRNA-1 and siRNA-2 did not consistently reduce ERK protein phosphorylation and did not consistently increase GSK3 protein expression (Figure 3b and Supplementary Figure S3B). We next cotransfected HEK293 primary embryonic kidney cells, which do not express endogenous N-Myc protein, with a construct expressing empty vector or SIRT2, together with a construct expressing empty vector, S62 mutant (S62A) N-Myc, T58 mutant (T58A) N-Myc, or wild-type N-Myc. Immunoblot analysis showed that overexpression of SIRT2 consistently upregulated the expression of T58-phosphorylated N-Myc and total N-Myc protein in cells transfected with S62 mutant N-Myc or wild-type N-Myc, but not in cells transfected with T58 mutant N-Myc (Supplementary Figure S3C). These data suggest that SIRT2 stabilizes N-Myc and c-Myc proteins without modulating ERK protein phosphorylation and GSK3 protein expression, and that SIRT2 stabilizes Myc protein downstream of Myc protein phosphorylation at T58.

SIRT2 stabilizes Myc proteins without modulating ERK protein phosphorylation and GSK3 protein expression. (a and b) BE(2)-C and MiaPaca-2 cells were transfected with scrambled control siRNA, SIRT2 siRNA-1 or SIRT2 siRNA-2, followed by protein extraction. (a) The expression of total N-Myc/c-Myc protein, N-Myc/c-Myc protein phosphorylated at S62 (S62-phos) and N-Myc/c-Myc protein phosphorylated at T58 (T58-phos) was analyzed by immunoblot with specific antibodies. (b) The expression of GSK3 protein, total ERK protein and phosphorylated ERK protein (phos-ERK) was analyzed by immunoblot with specific antibodies
SIRT2 represses NEDD4 gene transcription by directly binding to NEDD4 gene promoter and repressing NEDD4 promoter activity
The class III HDAC SIRT2 directly deacetylates histone H4K16,18 and could therefore directly repress gene transcription. To identify transcriptional target genes potentially responsible for SIRT2-induced Myc protein stabilization, we performed differential gene expression studies with Affymetrix gene array in BE(2)-C cells 30 h after transfection with scrambled control siRNA or SIRT2 siRNA-1. As shown in Supplementary Tables S3 and S4, the gene most significantly reactivated by SIRT2 siRNA-1 was the ubiquitin–protein ligase NEDD4. To validate the Affymetrix gene array data, we performed real-time RT-PCR and immunoblot analyses of NEDD4 expression in BE(2)-C and CHP134 neuroblastoma, and MiaPaca-2 and BXPC3 pancreatic cancer cells after transfection with control siRNA, SIRT2 siRNA-1 or SIRT2 siRNA-2, or in BE(2)-C and MiaPaca-2 cells after transfection with a construct expressing empty vector or SIRT2. As shown in Figure 4a and Supplementary Figure S4, the expression of NEDD4 was upregulated by SIRT2 siRNA-1 and SIRT2 siRNA-2 in BE(2)-C, MiaPaca-2 (Figure 4a), CHP134, and BXPC3 (Supplementary Figure S4A) cells, and the expression of NEDD4 was downregulated by a SIRT2 overexpression construct in BE(2)-C and MiaPaca-2 cells (Supplementary Figure S4B). Consistently, repression of SIRT2 activity with the SIRT2 inhibitor AC-93253 or Salermide also reactivated NEDD4 expression (Figure 4b and Supplementary Figure S5). These data demonstrate that NEDD4 is transcriptionally repressed by SIRT2, and that SIRT2 inhibitors can be applied to reverse the effect.

SIRT2 represses NEDD4 gene transcription by directly binding to NEDD4 gene promoter and repressing NEDD4 promoter activity. (a and b) BE(2)-C and MiaPaca-2 cells were transfected with scrambled control, SIRT2 siRNA-1 or SIRT2 siRNA-2 (a), or treated with vehicle control or 0.6 μM AC-93253 (b). NEDD4 mRNA and protein expression was analyzed by real-time RT-PCR and immunoblot. (c) ChIP assays were performed in BE(2)-C and MiaPaca-2 cells with a control, anti-SIRT2 or anti-acetyl H4K16 Ab, and real-time PCR with primers targeting control region (−2500 bp upstream of the NEDD4 gene transcription start site) or primers targeting part of the NEDD4 gene core promoter region (−394 to −263 bp upstream of the NEDD4 gene transcription start site). Fold enrichment of NEDD4 gene core promoter by control, anti-SIRT2 or anti-acetyl H4K16 Ab was calculated by dividing PCR products from primers targeting the NEDD4 gene core promoter by PCR products from primers targeting control region. Fold enrichment by control Ab was artificially set as 1.0. (d) BE(2)-C and MiaPaca-2 cells were transfected with scrambled control siRNA or SIRT2 siRNA-2 for 48 h, followed by ChIP assays with an anti-acetyl H4K16 Ab and real-time PCR with primers targeting control region or primers targeting the NEDD4 gene core promoter region. Fold change in acetylated H4K16 at NEDD4 gene core promoter was obtained after dividing fold enrichment of acetylated H4K16 at NEDD4 gene core promoter in SIRT2 siRNA-2-transfected samples by fold enrichment of acetylated H4K16 at NEDD4 gene core promoter in control siRNA-transfected samples. (e) BE(2)-C cells were transfected with control siRNA or SIRT2 siRNA-2 for 24 h, followed by transfection with a wild-type or a mutant NEDD4 gene promoter construct for another 24 h. Luciferase activity was measured and expressed as relative luciferase activity (wild-type NEDD4 promoter construct/mutant NEDD4 promoter construct). Error bars represented standard error. *P<0.05
We have previously shown that the histone deacetyase HDAC1, HDAC2, and SIRT1 repress gene transcription by binding to target gene promoters.6, 22, 24 We therefore performed chromatin immunoprecipitation (ChIP) assays with an anti-SIRT2 antibody (Ab), an anti-acetylated histone H4 lysine 16 (acetyl H4K16) Ab or a control Ab and PCR with primers targeting upstream control region or core promoter region of the NEDD4 gene promoter. The ChIP assays showed that the anti-SIRT2 and the anti-acetyl H4K16 antibodies efficiently immunoprecipitated the region of NEDD4 gene core promoter in BE(2)-C and MiaPaca-2 cells (Figure 4c), and that knocking down SIRT2 expression with SIRT2 siRNA-2 increased the presence of acetyl H4K16 at the NEDD4 gene core promoter by ∼2-fold in BE(2)-C and MiaPaca-2 cells (Figure 4d). To confirm that SIRT2 repressed NEDD4 gene promoter activity, we transfected BE(2)-C cells with control siRNA or SIRT2 siRNA-2 for 24 h, followed by transfection with a luciferase reporter construct carrying wild-type or mutant NEDD4 gene core promoter for another 24 h. Luciferase assays showed that repression of SIRT2 expression significantly activated the wild-type promoter, compared with the mutant NEDD4 gene promoter (Figure 4e). The data suggest that SIRT2 represses NEDD4 gene transcription by directly binding to NEDD4 gene core promoter, deacetylating histone H4K16 at NEDD4 gene core promoter, and represses NEDD4 gene transcription.
Repression of NEDD4 gene expression contributes to SIRT2-induced upregulation of Myc proteins
As a HECT ubiquitin–protein ligase, NEDD4 directly binds to substrate proteins and targets them for ubiquitination, leading to proteasome-mediated substrate protein degradation.25, 26 We therefore examined whether NEDD4 reduced Myc protein expression. As shown in Figure 5a, knocking down NEDD4 expression by NEDD4 siRNA reduced the expression of NEDD4 mRNA and protein, and increased the expression of N-Myc and c-Myc protein, but not N-Myc and c-Myc mRNA, in BE(2)-C and MiaPaca-2 cells. Importantly, cotransfection with SIRT2 siRNA blocked NEDD4 siRNA-mediated N-Myc and c-Myc protein upregulation (Figure 5a). We next cotransfected SHEP neuroblastoma cells, which do not express N-Myc or c-Myc oncoprotein, with an empty vector, N-Myc-overexpressing construct,24 and/or NEDD4-overexpressing construct.27 Immunoblot analysis revealed that transfection with the NEDD4 expression construct reduced N-Myc protein expression (Figure 5b). These data suggest that SIRT2-modulated transcriptional repression of NEDD4 leads to the upregulation of Myc proteins.

Repression of NEDD4 gene expression contributes to SIRT2-induced upregulation of Myc proteins. (a) BE(2)-C and MiaPaca-2 cells were transfected with scrambled control siRNA, NEDD4 siRNA or SIRT2 siRNA-1. NEDD4, N-Myc and c-Myc gene, and protein expression were analyzed by real-time RT-PCR and immunoblot. Error bars represented standard error. ***P<0.001. (b) SHEP neuroblastoma cells, which do not express N-Myc and c-Myc protein, were transfected with an empty vector, N-Myc-expressing construct and/or NEDD4-expressing construct. N-Myc protein expression was analyzed by immunoblot
NEDD4 reduces N-Myc protein expression by targeting N-Myc protein for ubiquitination
Next, we investigated whether NEDD4 directly binds to N-Myc and targets N-Myc for ubiquitination. As Myc oncoproteins are nuclear proteins and NEDD4 protein can be found in both the cytoplasm and the nucleus,28 we separated cellular protein from BE(2)-C and MiaPaca-2 cells into cytoplasmic and nuclear fractions. Immunoblot analyses revealed that SIRT2 and NEDD4 proteins localized in both the cytoplasm and the nucleus (Figure 6a and Supplementary Figure S6), indicating the possibility of NEDD4 protein binding to N-Myc protein in the nucleus. To demonstrate that N-Myc and NEDD4 form a protein complex, we transfected human embryonic HEK293 cells with Flag-tagged empty vector, an N-Myc-expressing construct, and/or a NEDD4-expressing construct; we then extracted protein from the cells and performed protein co-immunoprecipitation (IP) assays. Results showed that anti-N-Myc Ab could efficiently co-IP NEDD4 protein (Figure 6b). To demonstrate that the binding of NEDD4 to N-Myc leads to N-Myc protein ubiquitination in cells, we transfected HEK293 cells with a Flag-tagged empty vector, a HA-tagged ubiquitin expressing construct, a Flag-tagged N-Myc expressing construct, and/or a Flag-tagged NEDD4 expressing construct. Protein co-IP experiments with an anti-N-Myc Ab and immunoblot with an anti-HA Ab showed that protein ladders and smears representing polyubiquitinated N-Myc (N-Myc-UBn) were detectable by immunoblot, and that NEDD4 dramatically enhanced N-Myc protein ubiquitination (Figure 6c). These data suggest that NEDD4 protein binds to N-Myc protein and negatively regulates N-Myc protein stability by increasing its ubiquitination.

NEDD4 protein directly binds to N-Myc protein and targets N-Myc protein for ubiquitination. (a) BE(2)-C cells were transfected with control siRNA, NEDD4 siRNA or SIRT2 siRNA for 48 h. Cytoplasmic and nuclear protein was separated and analyzed by immunoblot with anti-NEDD4, anti-SIRT2, anti-GAPDH (marker for cytoplasmic protein), and anti-E2F1 (marker for nuclear protein) antibodies. (b) HEK293 cells were transfected with constructs expressing empty vector, N-Myc and/or NEDD4. Protein from the cells was immunoprecipitated with an anti-N-Myc Ab or a control mouse Ab, and IPproducts were analyzed by immunoblot with an anti-NEDD4 Ab. (c) HEK293 cells were transfected with constructs expressing HA-ubiquitin, Flag-N-Myc and/or Flag-NEDD4. Upper panel: protein from the cells was immunoprecipitated with an anti-N-Myc Ab, and co-IP products were analyzed by immunoblot with an anti-HA Ab. Lower panel: 5% of whole-cell lysates was used as input and probed with an anti-Flag Ab. (d) HEK293 cells were transfected with constructs expressing Flag-N-Myc and Flag-NEDD4. Protein from the cells was precipitated with anti-Flag M2 beads and incubated with E1, a panel of E2 enzymes, HA-ubiquitin (HA-Ub), and ATP. Polyubiquitinated protein was detected by immunoblot (IB) with an anti-HA Ab (upper panel). The indicated smear appearing above 110 kDa represented polyubiquitinated NEDD4 and N-Myc (N-Myc-UBn), and the smear/ladder between 76 and 110 kDa represented N-Myc-UBn. Immunoblot results with an anti-Flag M2 Ab showing Flag-tagged products are shown in the lower panel. (e) HEK293 cells were transfected with constructs expressing Flag-N-Myc, Flag-NEDD4, mutant Flag-NEDD4, or empty vector. Protein from the cells was precipitated with anti-Flag M2 beads, and precipitated protein was incubated with E1, UbcH5a, HA-ubiquitin, and ATP, followed by immunoblot analysis with an anti-HA Ab
To demonstrate that NEDD4 protein can directly target N-Myc protein for ubiquitination and to identify the E2 ubiquitin–protein ligases involved in the ubiquitination process, we performed in vitro ubiquitination assays. Flag-tagged N-Myc was incubated with recombinant human E1 enzyme, a panel of E2 enzymes, Flag-tagged NEDD4, HA-tagged ubiquitin, and ATP. As can be seen in Figure 6d, clear accumulation of N-Myc-UBn was observed with a specific family of E2 enzymes, UbcH5 (UbcH5a and UbcH5b). Other E2 enzymes tested produced little or no N-Myc polyubiquitination. In the reaction where wild-type NEDD4 was substituted with a mutated, catalytically inactive mutant NEDD4 (C894A) or empty vector, this N-Myc polyubiquitination was abolished (Figure 6e). Taken together, these experiments demonstrate that NEDD4 targets N-Myc protein for ubiquitination and degradation.
SIRT2 upregulates the expression of Aurora A
Aurora A interacts with N-Myc and blocks N-Myc protein degradation.8 While SIRT2 has recently been reported to exert tumor suppressor effects by inducing Aurora A protein degradation,29 our Affymetrix gene array data showed that Aurora A was one of the genes significantly downregulated by SIRT2 siRNA-1 in BE(2)-C cells (Supplementary Table S4). We therefore performed RT-PCR and immunoblot analysis of Aurora A expression in BE(2)-C and MiaPaca-2 cells after transfection with control siRNA, SIRT2 siRNA-1, or SIRT2 siRNA-2. Results showed that knocking down SIRT2 expression slightly reduced Aurora A mRNA but considerably reduced Aurora A protein expression (Figure 7a). We next treated BE(2)-C and MiaPaca-2 cells with vehicle control, the SIRT2 inhibitor AC-93253 at 0.8 μM or the SIRT1/SIRT2 inhibitor Salermide at 50 μM, at which Salermide inhibits SIRT2 but not SIRT1.21 RT-PCR and immunoblot analyses showed that inhibition of SIRT2 deacetylation activity with AC-93253 or Salermide significantly reduced Aurora A mRNA and protein expression (Figures 7b and c). The data suggest that SIRT2 can stabilize Myc protein and exert tumorigenic effects partly through upregulating Aurora A expression.

SIRT2 upregulates Aurora A expression. (a) BE(2)-C and MiaPaca-2 cells were transfected with scrambled control siRNA, SIRT2 siRNA-1 or SIRT2 siRNA-2 for 48 h, followed by real-time RT-PCR and immunoblot analyses of Aurora A mRNA and protein expression. (b and c) BE(2)-C and MiaPaca-2 cells were treated with vehicle control, the SIRT2 inhibitor AC-93253 at 0.8 μM (b), or Salermide at 50 μM (c) for 48 h, followed by real-time RT-PCR and immunoblot analyses of Aurora A mRNA and protein expression. Error bars represented standard error. *P<0.05
Discussion
Recent studies demonstrate that Myc oncoproteins can upregulate protein expression without modulating gene transcription by enhancing ribosome biogenesis, the formation of 7-methylguanosine caps on mRNAs and mRNA translation to protein.14, 15 In this study, we have shown that N-Myc and c-Myc oncoproteins upregulate the expression of SIRT2 protein, but not mRNA, in N-Myc overexpressing neuroblastoma and c-Myc overexpressing pancreatic cancer cells. The data suggest that Myc oncoproteins upregulate the expression of SIRT2 through a post-transcriptional mechanism, possibly by enhancing SIRT2 protein synthesis.
This study demonstrates that knocking down SIRT2 gene expression with siRNAs or repressing SIRT2 deacetylation activity with the SIRT2 inhibitor AC-93253 or Salemide reduces BE(2)-C neuroblastoma and MiaPaca-2 pancreatic cancer cell proliferation without inducing cell death. As SIRT2 is known to block programmed cell death by deacetylating p53 protein,30 we hypothesize that repression of SIRT2 does not induce significant cell death in BE(2)-C and MiaPaca-2 cells because p53 is mutated in the two cell lines.6, 24
One surprising finding of this study is that knocking down SIRT2 gene expression with siRNAs considerably reduces N-Myc protein expression in neuroblastoma cells and c-Myc protein expression in pancreatic cancer cells, but shows no effect on N-Myc and c-Myc mRNA expression. Consistently, the SIRT2 inhibitor AC-93253 and Salermide decrease the expression of N-Myc and c-Myc protein, but not mRNA. We have previously shown that the proteasome inhibitor MG-132 does not increase Myc protein expression in cancer cells transfected with Myc siRNAs, which ablates Myc mRNA.22 Here we show that MG-132 significantly increases Myc protein expression in cancer cells transfected with SIRT2 siRNAs, the repression of SIRT2 significantly decreases N-Myc and c-Myc protein half-life, and SIRT2 has no effect on the expression of phosphorylated ERK and GSK3 proteins. We conclude that SIRT2 upregulates Myc protein expression through blocking Myc protein degradation without modulating ERK protein phosphorylation and GSK3 protein expression.
This study has identified the ubiquitin–protein ligase NEDD4 as the gene most significantly repressed by SIRT2, and verified that NEDD4 gene expression can be reactivated by the SIRT2 deacetylation inhibitor AC-93253 and Salermide. Importantly, repression of NEDD4 with siRNA upregulates and overexpression of NEDD4 reduces the expression of N-Myc and c-Myc protein, but not mRNA. These data indicate that SIRT2 stabilizes N-Myc and c-Myc protein in neuroblastoma and pancreatic cancer cells at least partly by repressing NEDD4 gene transcription.
We have previously demonstrated that histone deacetyases HDAC1, HDAC2, and SIRT1 repress gene transcription by binding to target gene core promoters.6, 22, 24 Our present study shows that SIRT2 represses NEDD4 gene expression in neuroblastoma and pancreatic cells, that SIRT2 binds to NEDD4 gene core promoter and SIRT2 siRNA increases the presence of acetylated histone H4K16 at NEDD4 gene promoter, and that SIRT2 siRNA enhances NEDD4 promoter activity. These data suggest that SIRT2 represses NEDD4 gene transcription by direct binding to NEDD4 gene core promoter region, deacetylating histone H4K16 and, repressing NEDD4 gene promoter activity.
Myc oncoproteins are well known to be degraded through the ubiquitin–protein ligases Fbxw7, which degrades Myc protein after its phosphorylation at T58 by GSK3,31 and Skp2, which interacts with a non-phospho-dependent binding site of Myc oncoproteins.32, 33 Recently, the HectH9/Huwe1 ubiquitin–protein ligase has been shown to degrade N-Myc protein during neuronal differentiation.34 In the current study, we have identified the ubiquitin–protein ligase NEDD4, which is a member of the HECT ubiquitin–protein ligase family,25, 26 as a novel regulator of Myc protein ubiquitination and degradation. Our data demonstrate that NEDD4 directly binds to N-Myc protein, and targets N-Myc protein for ubiquitination by the ubiquitin conjugating enzymes (E2) UbcH5a and UbcH5b. Our findings therefore reveal a novel pathway through which Myc oncoproteins are ubiquitinated for proteasome-mediated degradation.
Aurora A binds to N-Myc and blocks N-Myc protein degradation.8 Kim et al.29 have recently reported that SIRT2 indirectly enhances the activity of anaphase-promoting complex/cyclosome (APC/C), a multi-subunit member of the RING finger family of ubiquitin ligases, leading to Aurora A protein ubiquitination and degradation. In contrast, in this study, we have confirmed that knocking down SIRT2 gene expression with two independent siRNAs and suppressing SIRT2 deacetylation activity with AC-93253 or Salermide consistently reduces Aurora A mRNA and protein expression in neuroblastoma and pancreatic cancer cells. While the mechanisms for the discrepancy between Kim et al.’s finding29 and our finding are unknown, we hypothesize that SIRT2 can either reduce or increase Aurora A expression, depending on whether SIRT2 predominantly interacts with APC/C or predominantly mediates Aurora A gene expression. Our own data suggest that SIRT2 can stabilize Myc oncoproteins and exert oncogenic effects by upregulating Aurora A expression.
The role of SIRT2 in tumorigenesis is currently controversial. An early study suggests that downregulation of SIRT2 confers glioma cell resistance to microtubule inhibitors, such as nocodazole.35 However, a recent study demonstrates that knocking down SIRT2 expression results in caspase 3-dependent apoptosis in glioma cells.36 Furthermore, a number of other recent papers demonstrate that SIRT2 is an oncogenic factor and a novel target for cancer therapy. SIRT2 mRNA and protein expression is significantly upregulated in primary acute myeloid leukemia (AML) blasts compared to counterpart normal cells from healthy individuals, and SIRT2 induces leukemia cell proliferation and resistance to apoptosis.37 SIRT2-deficient cells display increased susceptibility to apoptotic cell death induced by oxidative stress and the chemotherapeutic drug cisplatin and staurosporine.38 Although FoxO1 induces cell death and shows tumor suppressor activity, its function as a tumor suppressor is abrogated when it is bound and deacetylated by SIRT2.39 Importantly, small molecule SIRT2 inhibitors have unanimously shown anticancer effects. For example, the SIRT2-selective inhibitor AC-93253 induces apoptosis in cancer cells originating from various organs,20 and a number of 1,2-dihydrobenzochromen-derived SIRT2-selective inhibitors cause apoptosis and/or differentiation in leukemic cells.19 Additionally, the pan-SIRT inhibitor nicotinamide suppresses growth of carcinogen-induced mouse and human bladder cancer by reducing the deacetylation activity of SIRT2.40 Consistent with literature,41, 22, 42, 43, 44, 45 our data suggest that repression of SIRT2 alone or simultaneous repression of SIRT1 and SIRT2 with small molecule inhibitors, such as AC-93253, Salermide, Cambinol, and Tenovin-6, could be an effective strategy for the prevention and therapy of Myc-induced neuroblastoma and pancreatic cancer, and possibly other Myc-induced malignancies.
In summary, this study demonstrates that a novel pathway, involving post-transcriptional upregulation of SIRT2, repression of NEDD4, upregulation of Aurora A, and consequent decreased Myc protein ubiquitination contributes to N-Myc and c-Myc oncoprotein stability, neuroblastoma, and pancreatic cancer cell proliferation. Moreover, the SIRT2 inhibitor AC-93253 and Salermide upregulate NEDD4 expression, decrease Aurora A expression, reduce N-Myc and c-Myc protein expression, and induce neuroblastoma and pancreatic cancer cell growth inhibition. Our findings therefore identify upregulation of SIRT2 and repression of NEDD4 as important cofactors for Myc oncogenesis, and provide important evidence for the potential application of SIRT2 inhibitors for the prevention and therapy of Myc-induced neuroblastoma and pancreatic cancer.
Materials and Methods
Cell culture
Neuroblastoma BE(2)-C, CHP134, pancreatic cancer MiaPaca-2 cells, primary embryonic kidney HEK293 cells were cultured in RPMI or Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum.
siRNA and plasmid transfection
Cells were transfected with siRNAs from Qiagen (Hamburg, Germany) or Ambion (Austin, TX, USA) or plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) reagent with the protocol we described previously.6, 46 The sequences of siRNA targets are: 5′-CCCGGACGAAGATGACTTCTA-3′ for N-Myc siRNA-1; 5′-CGTGCCGGAGTTGGTAAAGAA3′ for N-Myc siRNA-2; 5′-CAGCGCGTTTCTTCTCCTGTA-3′ for SIRT2 siRNA-1; 5′-AATCTCCACATCCGCAGGCAT-3′ for SIRT2 siRNA-2; 5′-ATGGAGTTGATTAGATTACAA-3′ for NEDD4 siRNA.
RT-PCR and immunoblot analyses
Gene expression in tumor cells was examined by quantitative real-time RT-PCR as we described previously.22 For the analysis of protein expression by immunoblot, cells were lysed for either total cellular protein extraction with RIPA buffer or for nuclear/cytoplasmis protein fractionation with Nuclear Protein Extraction kit (Pierce, Rockford, IL, USA). After gel electrophoresis and western transfer, membranes were probed with mouse anti-N-Myc (1 : 1000), mouse anti-c-Myc (1 : 1000), mouse anti-SIRT2 (1 : 1000) (all from Santa Cruz Biotech, Santa Cruz, CA, USA), rabbit anti-NEDD4 (1 : 200), rabbit anti-Aurora A (1 : 500) (from Cell Signaling, Danvers, MA, USA), mouse anti-phosphorylated ERK (1 : 1000), rabbit anti-total ERK (1 : 1000) (both from Millipore, Billerica, MA, USA), mouse anti-total GSK3, rabbit anti-S62 phosphorylated c-Myc (N-Myc) (1 : 1000) (from Bethyl Laboratories, Montgomery, TX, USA) or rabbit anti-T58 phosphorylated c-Myc (N-Myc) Ab (Abcam, Cambridge, MA, USA), followed by horseradish peroxidase-conjugated anti-mouse (1 : 10 000) or anti-rabbit (1 : 20 000) antiserum (Santa Cruz Biotech). Protein bands were visualized with SuperSignal (Pierce). The membranes were lastly re-probed with an anti-actin Ab (Sigma, St Louis, MO, USA) as loading controls.
Affymetrix gene array study
BE(2)-C neuroblastoma cells were transfected with scrambled control siRNA or SIRT2 siRNA-1. Thirty hours after transfection, RNA was extracted from the cells with RNeasy mini kit (Qiagen). Differential gene expression was examined with Affymetrix GeneChip Gene 1.0 ST Arrays (Affymetrix, Santa Clara, CA, USA), according to the manufacturer’s instruction. Results from the microarray hybridization were analyzed with GeneSpring software (GeneSpring, Santa Clara, CA, USA) as we described.22
Alamar blue assays
Cell proliferation was examined with Alamar blue assays.47 Briefly, cells were plated into 96-well plates, transfected with various siRNAs or treated with different dosages of AC-93253 or Salermide in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum without antibiotics. Seventy-two hours later, cells were incubated with Alamar blue (Invitrogen) for 5 h, and plates were then read on a micro-plate reader at 570/595 nm. Results were calculated according to the optical density absorbance units and expressed as percentage change in cell number.
Cell cycle analysis
Seventy-two hours after siRNA transfection, cells were harvested and then resuspended at a concentration of 2 × 106 cells/ml in solution containing 2 μg/ml RNase (Sigma) and 50 μg/ml propidium iodide (Sigma). Cells were then run on FACScan (Becton Dickinson, Bedford, MA, USA), and cell cycle was analyzed with CellQuest software (Becton Dickinson).
ChIP assays
ChIP assays were performed with an anti-SIRT2 Ab, anti-acetyl H4K16 Ab or control mouse Ab and PCR with primers targeting upstream control region or core promoter region of NEDD4 gene promoter with the protocol we have described.24 Fold enrichment of NEDD4 gene promoter by the anti-SIRT2 Ab, anti-acetyl H4K16 Ab and control Ab was calculated by dividing the PCR product from NEDD4 gene core promoter region by the PCR product from upstream control region.
Luciferase assays
Modulation of NEDD4 gene promoter activity by SIRT2 was analyzed by luciferase assays. A wild-type NEDD4 gene core promoter construct was generated by cloning the NEDD4 gene core promoter region (−680 bp upstream to +212 bp downstream of the NEDD4 gene transcription start site) into a pLightSwitch_Prom construct (SwitchGear Genomics, Menlo Park, CA, USA). A mutant NEDD4 gene promoter construct was generated by cloning a DNA fragment −1335 to −392 bp upstream of the NEDD4 gene transcription start site. BE(2)-C neuroblastoma cells were transiently transfected with control siRNA or SIRT2 siRNA-2 for 24 h, followed by transfection with the wild-type or the mutant NEDD4 gene promoter construct for another 24 h. Luciferase activity was measured with Luciferase Assay System (SwitchGear Genomics) according to the manufacturer’s instructions and as described previously.6
Co-IP assays
Human embryonic HEK293 cells were transiently cotransfected with pShuttle empty vector or pShuttle-N-Myc24 together with pCDNA3.1-empty vector or pcDNA3.1-NEDD427 with Lipofectamine 2000 (Invitrogen) for 36 h. Cellular protein was then extracted and incubated overnight with 2 μg of a control or anti-N-Myc Ab (from Cell Signaling). Eluted proteins were immunoblotted with an anti-NEDD4 Ab.
In vivo ubiquitination assays
HEK293 cells were transiently cotransfected with pcDNA3.1-HA-ubiquitin plus pCDNA3.1-empty vector or pCDNA3.1-Flag-NEDD4 plus pShuttle empty vector or pShuttle-Flag-N-Myc with Lipofectamine 2000 (Invitrogen). Thirty-six hours after transfection, cells were treated with 30 μM MG-132 to preserve multiubiquitin chains of N-Myc. Three hours later, the cells were lysed under denaturing conditions to disrupt non-covalent interactions. Total cell lysates were then immunoprecipitated with 2 μg of anti-N-Myc Ab. Eluted proteins were immunoblotted with an anti-HA Ab (Santa Cruz Biotech).
In vitro ubiquitination assays
HEK293 cells were transfected with Flag-tagged N-Myc, NEDD4, mutant NEDD4 (C894A) or empty vector expression constructs. 36 h after transfection, cells were harvested in NP40 buffer (50 mM Tris-HCl, pH 8; 100 mM NaCl; 5 mM EDTA; 0.5% NP-40) with protease and phosphatase inhibitors. Three milligram of total protein was used for IP with anti-Flag M2 beads (Sigma), followed by several washes with in vitro ubiquitination buffer (25 mM Tris-HCl, pH 7.6, 5 mM MgCl2, 100 mM NaCl) containing protease and phosphatase inhibitors. Flag-tagged NEDD4, mutant NEDD4 (C894A), or empty vector were eluted from the beads using 3 × Flag peptide (Sigma). Flag-N-Myc protein bound to the beads were then incubated for 90 min at 32°C in in vitro ubiquitination buffer containing 100 ng of E1, 150 ng of E2 enzyme, eluted NEDD4, mutant NEDD4 (C894A), or empty vector, 5 μg of HA-Ub, 2 mM ATP and 2 mM DTT. After incubation, the samples were washed and resolved by SDS-PAGE and analyzed by western blot using anti-HA and anti-Flag antibodies.
Statistical analysis
All experiments were repeated for at least three times in duplicates. All data for statistical analysis were calculated as mean±S.E. Differences were analyzed for significance using ANOVA among groups or unpaired t-test for two groups. A probability value of 0.05 or less was considered significant.
Abbreviations
- Ab:
-
antibody
- acetyl H4K16:
-
acetylated histone H4 lysine 16
- ChIP:
-
chromatin immunoprecipitation
- ERK:
-
extracellular signal-regulated protein kinase
- GSK3:
-
glycogen synthase kinase 3
- H4K16:
-
histone H4 lysine 16
- HDAC:
-
histone deacetylase
- IP:
-
immunoprecipitation
- N-Myc-UBn:
-
polyubiquitinated N-Myc
- S62:
-
serine 62
- T58:
-
threonine 58
- T1/2:
-
half-life
References
Gustafson WC, Weiss WA . Myc proteins as therapeutic targets. Oncogene 2010; 29: 1249–1259.
Brodeur GM . Neuroblastoma: Biological insights into a clinical enigma. Nat Rev Cancer 2003; 3: 203–216.
Maris JM, Matthay KK . Molecular biology of neuroblastoma. J Clin Oncol 1999; 17: 2264–2279.
Mahlamaki EH, Barlund M, Tanner M, Gorunova L, Hoglund M, Karhu R et al. Frequent amplification of 8q24, 11q, 17q, and 20q-specific genes in pancreatic cancer. Genes Chromosomes Cancer 2002; 35: 353–358.
Schleger C, Verbeke C, Hildenbrand R, Zentgraf H, Bleyl U . C-myc activation in primary and metastatic ductal adenocarcinoma of the pancreas: Incidence, mechanisms, and clinical significance. Mod Pathol 2002; 15: 462–469.
Marshall GM, Gherardi S, Xu N, Neiron Z, Trahair T, Scarlett CJ et al. Transcriptional upregulation of histone deacetylase 2 promotes myc-induced oncogenic effects. Oncogene 2010; 29: 5957–5968.
Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR . Multiple ras-dependent phosphorylation pathways regulate myc protein stability. Genes Dev 2000; 14: 2501–2514.
Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N et al. Stabilization of n-myc is a critical function of aurora a in human neuroblastoma. Cancer Cell 2009; 15: 67–78.
Amati B . Myc degradation: Dancing with ubiquitin ligases. Proc Natl Acad Sci USA 2004; 101: 8843–8844.
Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J et al. Genomic targets of the human c-myc protein. Genes Dev 2003; 17: 1115–1129.
Patel JH, Loboda AP, Showe MK, Showe LC, McMahon SB . Analysis of genomic targets reveals complex functions of myc. Nat Rev Cancer 2004; 4: 562–568.
Pelengaris S, Khan M, Evan G . C-myc: More than just a matter of life and death. Nat Rev Cancer 2002; 2: 764–776.
Eilers M, Eisenman RN . Myc’s broad reach. Genes Dev 2008; 22: 2755–2766.
van Riggelen J, Yetil A, Felsher DW . Myc as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer 2010; 10: 301–309.
Cole MD, Cowling VH . Transcription-independent functions of myc: Regulation of translation and DNA replication. Nat Rev Mol Cell Biol 2008; 9: 810–815.
Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK . Histone deacetylases and cancer: Causes and therapies. Nat Rev Cancer 2001; 1: 194–202.
Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G et al. Loss of acetylation at lys16 and trimethylation at lys20 of histone h4 is a common hallmark of human cancer. Nat Genet 2005; 37: 391–400.
Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW et al. Sirt2 is a histone deacetylase with preference for histone h4 lys 16 during mitosis. Genes Dev 2006; 20: 1256–1261.
Rotili D, Carafa V, Tarantino D, Botta G, Nebbioso A, Altucci L et al. Simplification of the tetracyclic sirt1-selective inhibitor mc2141: Coumarin- and pyrimidine-based sirt1/2 inhibitors with different selectivity profile. Bioorg Med Chem 2011; 19: 3659–3668.
Zhang Y, Au Q, Zhang M, Barber JR, Ng SC, Zhang B . Identification of a small molecule sirt2 inhibitor with selective tumor cytotoxicity. Biochem Biophys Res Commun 2009; 386: 729–733.
Lara E, Mai A, Calvanese V, Altucci L, Lopez-Nieva P, Martinez-Chantar ML et al. Salermide, a sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene 2009; 28: 781–791.
Marshall GM, Liu PY, Gherardi S, Scarlett CJ, Bedalov A, Xu N et al. Sirt1 promotes n-myc oncogenesis through a positive feedback loop involving the effects of mkp3 and erk on n-myc protein stability. PLoS Genet 2011; 7: e1002135.
Gregory MA, Qi Y, Hann SR . Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem 2003; 278: 51606–51612.
Liu T, Tee AE, Porro A, Smith SA, Dwarte T, Liu PY et al. Activation of tissue transglutaminase transcription by histone deacetylase inhibition as a therapeutic approach for myc oncogenesis. Proc Natl Acad Sci USA 2007; 104: 18682–18687.
Yang B, Kumar S . Nedd4 and nedd4-2: Closely related ubiquitin-protein ligases with distinct physiological functions. Cell Death Differ 2010; 17: 68–77.
Rotin D, Kumar S . Physiological functions of the hect family of ubiquitin ligases. Nat Rev Mol Cell Biol 2009; 10: 398–409.
Harvey KF, Shearwin-Whyatt LM, Fotia A, Parton RG, Kumar S . N4wbp5, a potential target for ubiquitination by the nedd4 family of proteins, is a novel golgi-associated protein. J Biol Chem 2002; 277: 9307–9317.
Hamilton MH, Tcherepanova I, Huibregtse JM, McDonnell DP . Nuclear import/export of hrpf1/nedd4 regulates the ubiquitin-dependent degradation of its nuclear substrates. J Biol Chem 2001; 276: 26324–26331.
Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z, Xu X et al. Sirt2 maintains genome integrity and suppresses tumorigenesis through regulating apc/c activity. Cancer Cell 2011; 20: 487–499.
Li Y, Matsumori H, Nakayama Y, Osaki M, Kojima H, Kurimasa A et al. Sirt2 down-regulation in hela can induce p53 accumulation via p38 mapk activation-dependent p300 decrease, eventually leading to apoptosis. Genes Cells 2011; 16: 34–45.
Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN et al. The fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-myc protein degradation. Proc Natl Acad Sci USA 2004; 101: 9085–9090.
Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP . Skp2 regulates myc protein stability and activity. Mol Cell 2003; 11: 1177–1188.
von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C et al. The f-box protein skp2 participates in c-myc proteosomal degradation and acts as a cofactor for c-myc-regulated transcription. Mol Cell 2003; 11: 1189–1200.
Zhao X, Heng JI, Guardavaccaro D, Jiang R, Pagano M, Guillemot F et al. The hect-domain ubiquitin ligase huwe1 controls neural differentiation and proliferation by destabilizing the n-myc oncoprotein. Nat Cell Biol 2008; 10: 643–653.
Inoue T, Hiratsuka M, Osaki M, Yamada H, Kishimoto I, Yamaguchi S et al. Sirt2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene 2007; 26: 945–957.
He X, Nie H, Hong Y, Sheng C, Xia W, Ying W . Sirt2 activity is required for the survival of c6 glioma cells. Biochem Biophys Res Commun 2012; 417: 468–472.
Dan L, Klimenkova O, Klimiankou M, Klusmann JH, van den Heuvel-Eibrink MM, Reinhardt D et al. The role of sirtuin 2 activation by nicotinamide phosphoribosyltransferase in the aberrant proliferation and survival of myeloid leukemia cells. Haematologica 2012; 97: 551–519.
Matsushita N, Takami Y, Kimura M, Tachiiri S, Ishiai M, Nakayama T et al. Role of nad-dependent deacetylases sirt1 and sirt2 in radiation and cisplatin-induced cell death in vertebrate cells. Genes Cells 2005; 10: 321–332.
Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S et al. Cytosolic foxo1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol 2011; 12: 665–675.
Kim WJ, Lee JW, Quan C, Youn HJ, Kim HM, Bae SC . Nicotinamide inhibits growth of carcinogen induced mouse bladder tumor and human bladder tumor xenograft through up-regulation of runx3 and p300. J Urol 2011; 185: 2366–2375.
Marks PA, Breslow R . Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol 2007; 25: 84–90.
Heltweg B, Gatbonton T, Schuler AD, Posakony J, Li H, Goehle S et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res 2006; 66: 4368–4377.
Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 2008; 13: 454–463.
Li L, Wang L, Wang Z, Ho Y, McDonald T, Holyoake TL et al. Activation of p53 by sirt1 inhibition enhances elimination of cml leukemia stem cells in combination with imatinib. Cancer Cell 2012; 21: 266–281.
Yuan H, Wang Z, Li L, Zhang H, Modi H, Horne D et al. Activation of stress response gene sirt1 by bcr-abl promotes leukemogenesis. Blood 2012; 119: 1904–1914.
Tee AE, Marshall GM, Liu PY, Xu N, Haber M, Norris MD et al. Opposing effects of two tissue transglutaminase protein isoforms in neuroblastoma cell differentiation. J Biol Chem 2010; 285: 3561–3567.
Liu T, Liu PY, Tee AE, Haber M, Norris MD, Gleave ME et al. Over-expression of clusterin is a resistance factor to the anti-cancer effect of histone deacetylase inhibitors. Eur J Cancer 2009; 45: 1846–1854.
Acknowledgements
We thank Dr. Xuejun Jiang for NEDD4 expression construct. This work was supported, in part, by a Cancer Council New South Wales (Australia) project grant (TL, AVB, and CJS) and a National Institutes of Health (USA) project grant (TL). TL is a recipient of an ARC Future Fellowship; AVB and CJS are recipients of Cancer Institute New South Wales Fellowships; and SK is a recipient of a National Health and Medical Research Council (Australia) Senior Principal Research Fellowship (1002863). Children’s Cancer Institute Australia is affiliated with University of New South Wales and Sydney Children’s Hospital.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by H Ichijo
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Rights and permissions
About this article
Cite this article
Liu, P., Xu, N., Malyukova, A. et al. The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death Differ 20, 503–514 (2013). https://doi.org/10.1038/cdd.2012.147
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2012.147
Keywords
This article is cited by
-
The role of NEDD4 related HECT-type E3 ubiquitin ligases in defective autophagy in cancer cells: molecular mechanisms and therapeutic perspectives
Molecular Medicine (2023)
-
Neuroblastoma and the epigenome
Cancer and Metastasis Reviews (2021)
-
Ubiquitin-specific protease 7 is a druggable target that is essential for pancreatic cancer growth and chemoresistance
Investigational New Drugs (2020)
-
NEDD4 expression is associated with breast cancer progression and is predictive of a poor prognosis
Breast Cancer Research (2019)
-
Acetylation regulates ribonucleotide reductase activity and cancer cell growth
Nature Communications (2019)
