
Abstract
The glial environment is an important determinant of neuronal health in experimental models of neurodegeneration. Specifically, astrocytes have been shown, dependent on context, to be both injurious and protective. Human pluripotent stem cells offer a powerful new system to improve our understanding of the mechanisms underlying astrocyte-mediated neuroprotection. Here, we describe a human embryonic stem cell (HESC)-based system to assess the scope and mechanism of human astrocyte-mediated neuroprotection. We first report the generation of enriched and functional HESC-derived astrocytes, by combining BMP-mediated Smad and LIF-mediated JAK-STAT signalling. These astrocytes promote the protection of HESC-derived neurons against oxidative insults. Moreover, their neuroprotective capacity can be greatly enhanced by treatment with the nuclear factor-erythroid 2-related factor 2 (Nrf2)-activating triterpenoid 1[2-Cyano-3,12-dioxool-eana-1,9(11)-dien-28-oyl] trifluoroethylamide (CDDOTFEA). Activation of the transcription factor Nrf2 in human astrocytes by CDDOTFEA treatment induced expression of the glutamate-cysteine ligase (GCL) catalytic subunit, leading to enhanced GCL activity and glutathione production, and strong neuroprotection against H2O2. This enhanced neuroprotection was found to be dependent on astrocytic GCL activity, unlike the basal neuroprotection afforded by untreated astrocytes. Direct treatment of HESC-derived neurons with CDDOTFEA elicited no induction of Nrf2 target genes, nor any neuroprotection. Thus, human astrocytes can mediate neuroprotection through glutathione-dependent and glutathione-independent mechanisms, and represent a therapeutic target for human disorders associated with neuronal oxidative stress.
Similar content being viewed by others


Main
Neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis (ALS) are untreatable conditions that collectively represent a major healthcare burden. Improved understanding of the biology of these diseases is required in order to develop neuroprotective and ultimately reparative treatments. Until recently, neurodegenerative diseases have been largely regarded as exclusively neuronal disorders. However, recent findings have challenged this concept, implicating non-cell-autonomous mechanisms of neurodegeneration mediated by astrocytes in acute and chronic disorders.1, 2
The role of astrocytes in influencing neuronal viability has primarily been characterised in rodent systems. Advances in human stem cell biology now allow the modelling of human astrocyte–neuronal interaction under physiological and injury paradigms. Although there are robust platforms for the generation of neural stem cells and functional neurons, the generation of enriched and functional human astrocytes is comparatively understudied.3, 4 While our understanding of astrogliogenesis remains incomplete, it is well established that neurogenesis precedes gliogenesis and this in turn is reflected in differential temporal phenotypic potential of neural precursor cells (NPCs); young precursors are neuron restricted while aged precursors are glial competent.5, 6, 7 In addition to poorly understood epigenetic changes that control temporal competence to astrogliogenesis, extrinsic signals are important determinants of astrocyte fate, including the JAK-STAT pathway activated by cytokines such as LIF, CNTF, and cardiotrophin 1 as well as BMP signalling acting through the Smad transcription factor family.8, 9 Synergistic interaction between these two signalling pathways raises the possibility that dual activation of STAT and BMP signalling will promote astrogliogenesis from human ES-derived neural precursors.10
While the underlying cause of individual neurodegenerative diseases is unknown, multiple lines of evidence implicate oxidative stress in disease pathogenesis.11 The support of neuronal antioxidant defences by astrocytes represents a key neuroprotective role and its manipulation represents an emerging therapeutic strategy for a range of acute and chronic neurological disorders associated with oxidative stress.12 One attractive target is the transcription factor nuclear factor-erythroid 2-related factor 2 (Nrf2), a master regulator of antioxidant defences and drug-metabolising enzymes. The binding of Nrf2 to a cis-acting DNA promoter sequence called the antioxidant response element (ARE), allows trans-activation of a group of genes that includes key components of the glutathione antioxidant system.13 Under normal conditions, Nrf2 is bound in the cytoplasm by Kelch-like ECH-associated protein 1 (Keap1) and targeted for proteasomal degradation.14 However, the Keap1-dependent turnover of Nrf2 can be inhibited by oxidative stress and a variety of small molecules, leading to an accumulation of Nrf2 whereupon it accumulates in the nucleus and activates ARE-driven genes. Activation of Nrf2 by mild oxidative stress in astrocytes contributes to neuroprotective ischaemic preconditioning,15 although other mechanisms exist.16 Furthermore, pharmacological activation of astrocytic Nrf2, or overexpression of Nrf2 in astrocytes, confers a neuroprotective response in rodent models of Parkinson's disease, and ALS.17, 18, 19 The mechanism behind this non-cell-autonomous neuroprotection is thought to be through the upregulation of astrocytic glutathione biosynthesis and secretion, breakdown products of which are used by the neurons to support their own glutathione levels.18 Noting inter-species functional differences, the relevance of these rodent-based astrocyte-mediated neuroprotective effects to human experimental systems is unknown.20
Here, we describe a novel protocol for generating highly enriched, functional astrocytes derived from human embryonic stem cells (HESCs), using a combination of developmental cues. By generating a separate population of neurons derived from the same HESC line, we establish a human-centred platform for assessing the neuroprotective properties of compounds when administered either directly to neurons, or in triggering a non-cell-autonomous neuroprotective response in astrocytes. As proof-of-concept, we demonstrate a key capacity for human astrocytes to mediate neuroprotective responses, including those triggered by a small molecule activator of Nrf2 that can only act in this non-cell-autonomous manner.
Results
Generation of enriched and functional astrocytes from human ES cells
In order to establish a platform to study astrocyte–neuronal interactions, we first generated a stable human neural precursor cell (hNPC) population. Using established protocols, HESC neural aggregates were first derived.21 At day 8, neural aggregates were mechanically dissociated and plated as an adherent monolayer in defined conditions on gelatin, selecting for neural rosette-like structures at each passage.22 By day 30, a near homogeneous population of hNPCs were obtained that were both nestin and 3CB2 positive (93.9±1.0% and 82.7±2.1%, respectively; Figures 1a and b) and had a bipolar morphology consistent with a radial glial identity.

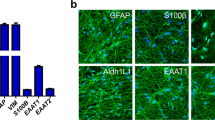
Derivation of astrocytes from HESCs. hNPC markers were quantified by immunohistochemistry for (a) nestin and (b) 3CB2-positive staining (scale bar, 100 μm) and analysed for (c) the presence of BMP and LIF signalling mediators by RT-PCR. hNPCs were treated with BMP2, BMP4, and LIF in combination and (d) mRNA expression of BLBP and GS analysed by quantitative real-time PCR, normalised to GAPDH. *P<0.05 ANOVA, Newman–Keuls post-test (n=3). Astrocyte yield was confirmed by (e–h) quantification of GFAP-positive immunostaining (scale bar, 100 μm), and (i–k) the presence of astrocyte markers AQP4, S100β and EAAT1 was examined to confirm astrocyte identity (scale bar, 50 μm). *P<0.05 ANOVA, Newman–Keuls post-test (n=3). (l) Astrocyte function was confirmed by examining sodium-dependent glutamate uptake. *P<0.05 paired t-test (n=3)
In order to generate astroglia, hNPCs were further propagated until day 55 when they expressed, along with nestin, and key receptors and downstream signalling effectors for astrogliogenesis. These included BMP and LIF receptors and, SMAD1 and STAT3, by RT-PCR (Figure 1c). Having previously shown that BMP4 promotes astrogliogenesis,5 we sought to refine this process in order to obtain more enriched astroglial populations for further enrichment. We systematically tested the efficacy of BMP4 alone and in combination with LIF and BMP2, which have been shown previously to promote astroglial commitment.9, 10 Astroglial specification was screened by measuring differential gene expression of brain lipid binding protein (BLBP), a neural precursor marker, and glutamine synthetase (GS), an astrocyte-expressed enzyme required for production of glutathione and glutamate recycling in neurotransmission. Gene expression analysis revealed downregulation of BLBP and upregulation of GS with all treatment groups relative to control (P<0.05). The combination of BMP4 and LIF resulted in the greatest change in gene expression (Figure 1d). Quantitative immunohistochemistry was next undertaken with astrocyte differentiation determined by downregulation of nestin and concomitant upregulation of GFAP staining, along with a change in morphology from bipolar to stellate. Although all treatment groups increased the numbers of cells with GFAP staining, the combination of BMP4 and LIF resulted in the greatest upregulation (88.4±2.0% versus control 35.8±3.6%; Figures 1e–h). Prolonged exposure (12 days) to BMP4 and LIF increased GFAP-positive staining to 95.7±3.1%. Furthermore, GFAP-positive astrocytes derived by BMP4/LIF co-treatment also stained positive for other markers of astrocyte differentiation; aquaporin 4 (79.4±1.0%) (Figure 1i), S100β (90.1±2.0%) (Figure 1j), and EAAT1 (89.5±3.2%) (Figure 1k). A defining physiological role of astrocytes is glutamate uptake mediated by Na+-dependent channels that include members of the excitatory amino-acid transporter family EAAT1 and EAAT2.23 Radiolabelled glutamate uptake assays revealed negligible hNPC uptake in contrast to robust Na+-dependent glutamate uptake by BMP4/LIF-derived astrocytes consistent with astrocyte function (Figure 1l). Combined BMP4/LIF treatment was therefore used for subsequent functional and neuroprotection experiments.
Human astrocytes attenuate oxidative neuronal injury
Having established functional hNPC-derived astrocytes, we next generated an enriched population of neurons from HESCs in order to examine human astrocyte–neuronal interaction in the context of oxidative stress. Neural stem cells were generated from H9 HESCs as described by Koch et al.22; following differentiation for 10 days 95.5±1.4% were TuJ1 positive upon immunostaining (Figure 2a). Studies predominantly based in rodent systems have shown that astrocytes can release soluble factors that support the growth, development, and survival of neurons, and so astrocyte-conditioned medium (ACM) can be neuroprotective.24 We therefore sought to determine whether medium conditioned by HESC-derived astrocytes could exert a neuroprotective effect on HESC-derived neurons, using H2O2 to model an oxidative insult (Figures 2b and c). Cell death in response to H2O2 treatment was quantified with the CellTiter-glo assay and confirmed by quantitative immunohistochemistry determining activated caspase-3/TuJ1 positive co-staining (Figures 2d–f). Addition of ACM resulted in a significant neuroprotective effect against H2O2-induced injury, compared with unconditioned medium (31.0±1.8% loss of viability compared with control: 41.4±1.4% loss of viability; Figure 2c, P<0.05). Immunofluorescent analysis of caspase-3 activation revealed similar results: treatment of neurons with 50 μM H2O2 caused an increase of active caspase-3-positive neurons from 1.5±0.7% to 43.6±5.0%; however, the presence of ACM reduced the level of H2O2-induced caspase-3 activation to 30.2±2.5% (Figures 2d–f). Thus, human astrocytes are able to protect neurons against oxidative insults through the release of soluble factors.

ACM protects neurons from peroxidative injury. Highly enriched HESC-derived neurons demonstrated (a) >95% TuJ1-positive immunostaining (scale bar, 100 μm). (b) HESC-derived astrocyte cultures were used to condition minimal medium over 24 h. (c) Enriched HESC-derived neuronal cultures were treated with H2O2 (50 μM) for 6 h in minimal medium and ACM, and neuronal viability quantified. (d–f) Representative TuJ1 and activated caspase-3 co-staining (scale bar, 50 μm). *P<0.05 ANOVA, Newman–Keuls post-test (n=3)
CDDOTFEA treatment reveals glutathione-dependent and glutathione-independent mechanisms for astrocyte-mediated neuroprotection
Nrf2 activation in rodent astrocytes can confer increased antioxidant capacity on neurons by mechanisms that include enhanced glutathione biosynthesis and secretion.15, 18, 19 There is also growing evidence for Nrf2-activating synthetic triterpenoids as potential therapeutic agents in experimental models of neurodegenerative diseases associated with oxidative stress.25 To test their relevance to human systems, we examined whether the triterpenoid CDDOTFEA26 could induce Nrf2 target gene expression in human astrocytes and whether this leads to an enhanced neuroprotective response.
We first studied the expression of two Nrf2 target genes, NFE2L2 (encoding Nrf2 itself) and GCLC (the glutamate-cysteine ligase catalytic subunit). The GCLC gene encodes the catalytic subunit of GCL, which performs the rate-limiting step in glutathione biosynthesis. Compared with neurons, astrocytes showed far higher basal levels of both NFE2L2 and GCLC, indicative of a higher basal Nrf2 activity (Figure 3a). Moreover, these levels were further increased by treatment of astrocytes with CDDOTFEA (Figure 3a). Consistent with an upregulation in GCLC expression, cell-free extracts taken from CDDOTFEA-treated astrocytes exhibited elevated levels of GCL enzyme activity (24 h, 46.8±18% greater than control) (Figure 3b). To determine whether this in turn translated to increased glutathione levels in astrocytes, we assessed intracellular glutathione content using an assay based on monochlorobimane (MCB). MCB is non-fluorescent in its native state and becomes fluorescent upon conjugation to glutathione in the cell.27 CDDOTFEA treatment of astrocytes resulted in a substantial increase in glutathione levels (6 h, 34.7±13.8% greater than control, P<0.05; 24 h, 54.4±17.7% greater than control, P<0.05; Figure 3c) that was completely blocked by co-treatment with the GCL inhibitor buthionine sulfoximine (BSO) (CDDOTFEA/BSO: −37.3±14.5% at 6 h, −65.8±6.3% at 24 h; BSO: −16.1±8.4% at 6 h, −63.0±3.8% at 24 h; Figure 3c). Thus, CDDOTFEA treatment of human astrocytes upregulates Nrf2 target gene expression, GCL enzyme activity, and glutathione production, raising the possibility that this influences their neuroprotective capacity.

Astrocytes upregulate NRF2 pathway with CDDOTFEA treatment. (a) mRNA expression of NFE2L2 and GCLC were examined by quantitative real-time PCR, normalised to GAPDH. Cell types examined included enriched human neuron and astrocyte cultures, and human astrocyte cultures treated with CDDOTFEA (250 nM) for 6 h. *P<0.05, **P<0.01 paired t-test (n=3). (b) GCL enzyme activity was examined in enriched human astrocyte cultures treated with CDDOTFEA (250 nM) for 6 h. *P<0.05 paired t-test (n=3). (c) Enriched human astrocytes were treated with CDDOTFEA (250 nM), BSO (200 μM), and CDDOTFEA + BSO for 6 h and 24 h, and intracellular GSH quantified by the MCB assay. *P<0.05 ANOVA, Newman–Keuls post-test (n=3)
Before investigating the capacity of CDDOTFEA to boost astrocyte-mediated neuroprotection in our human system, we first sought to establish its dependence on Nrf2 using cultures derived from wild-type and Nrf2-deficient mice. We first established that CDDOTFEA-mediated neuroprotection required the presence of astrocytes (Figure 4a). CDDOTFEA treatment protected neurons against H2O2-induced death in mixed Nrf2+/+ cortical cultures (90% NeuN-positive neurons, 10% GFAP-positive astrocytes;15, 25 however, no protection was observed in astrocyte-free Nrf2+/+ neuronal cultures (Figure 4a). We then investigated the Nrf2 dependence of CDDOTFEA-induced neuroprotection (Figure 4b). We found that CDDOTFEA-induced neuroprotection of mixed cultures was dramatically diminished in Nrf2−/− mixed cultures, compared with those from Nrf2+/+ mice. These data indicate that CDDOTFEA-mediated neuroprotection is indeed mediated through its effects on the Nrf2 pathway (Figure 4b).

CDDOTFEA drug treatment is neuroprotective by a GSH-dependent, astrocyte-mediated mechanism. (a) CDDOTFEA-induced neuroprotection requires the presence of astrocytes. Mixed mouse cortical cultures (90% NeuN+ neurons, 10% GFAP+ astrocytes) or astrocyte-free mouse neuronal cultures (>98% NeuN+, <0.2% GFAP+) were treated with CDDOTFEA (100 nM) for 24 h, before treatment with H2O2 (100 μM) for a further 24 h, after which cultures were fixed and levels of cell death assessed. *P<0.05 paired t-test (n=4). (b) CDDOTFEA-induced neuroprotection is Nrf2-dependent. CDDOTFEA-induced neuroprotection in mixed Nrf2+/+ or Nrf2−/− mouse cortical cultures was studied using the same protocols as (a). *P<0.05 paired t-test (n=4). (c) Enriched human astrocyte cultures were treated with CDDOTFEA (250 nM)±BSO (200 μM) for 6 h, the drug washed out, and fresh minimal medium replaced for conditioning over 24 h. ACM was derived as previously and supplemented with H2O2. (d) Human neuronal viability was assessed upon treatment with H2O2 (50 μM) for 6 h, in ACM derived from untreated and CDDOTFEA-treated enriched astrocytes. BSO co-treatment was used to block GCL activity, demonstrating GCL-dependent and -independent neuroprotective effects of ACM. (e–g) Representative TuJ1 and activated caspase-3 co-staining (scale bar, 50 μm). *P<0.05 ANOVA, Newman–Keuls post-test (n=3). (h) Human neuronal expression of NFE2L2 and GCLC mRNA with 6 h CDDOTFEA (250 nM) treatment were examined by quantitative real-time PCR. (i) The effect of treatment with CDDOTFEA (250 nM) for 24 h on human neuronal viability was determined over a range of H2O2 concentrations (0–200 μM, 6 h). *P<0.05 paired t-test (n=3)
To investigate whether CDDOTFEA treatment of human astrocytes can promote protection of human neurons, astrocytes were treated with or without CDDOTFEA for 6 h, in the presence or absence of BSO, after which all drugs were washed out and fresh medium added for the astrocytes to condition for 24 h (Figure 4c). Medium conditioned by CDDOTFEA-treated astrocytes was significantly more neuroprotective against oxidative injury than medium conditioned by untreated astrocytes as determined by viability assay and active caspase-3 immunohistochemistry (Figures 4d–g). The enhanced protective response induced by CDDOTFEA was blocked if GCL activity was inhibited by co-treatment with BSO (Figures 4d and g), implicating enhanced astrocytic GCL activity and glutathione production as the mechanism underlying protection conferred by CDDOTFEA treatment in contrast to a glutathione-independent protective effect mediated by basal untreated astrocytes (Figure 4d). We next examined whether CDDOTFEA treatment of neurons alone was also protective against oxidative injury. Unlike astrocytes, CDDOTFEA treatment of neurons failed to induce NFE2L2 or GCLC mRNA expression (1.25±0.2, 1.23±0.1 fold change) (Figure 4h), and importantly conferred no significant protective effect against H2O2-induced oxidative injury, at a range of concentrations (Figure 4i). These findings reveal that CDDOTFEA can only exert its human neuroprotective effects non-cell-autonomously via Nrf2 activation and glutathione production in astrocytes. Thus, basal and CDDOTFEA-induced astrocyte-mediated neuroprotection against oxidative injury involves distinct glutathione-dependent and glutathione-independent mechanisms.
Discussion
The role of the glial environment in modulating health, injury, and protection of neurons is becoming increasingly well recognised.1, 28 In order to further understand the relevance of glial signalling to human neuronal injury, there is a need to first establish appropriate human systems of glial–neuronal interaction. This study highlights the utility of HESCs for in-vitro human glial–neuronal modelling and demonstrates generation of highly enriched functional human astrocytes that promote survival of neurons following oxidative injury. In addition, we find that human astrocytic neuroprotective responses involve multiple mechanisms and can be manipulated by small molecules.
Previous studies have focussed predominantly on neuronal differentiation of HESCs with some modelling of isolated neuronal injury.1, 2 In contrast, the study of functional HESC-derived astrocytes is limited with negligible investigation of human glial–neuronal interaction. Astrocytic differentiation is determined temporally by intrinsic mechanisms5 and exogenous factors, including BMP4 and LIF;9 LIF and other members of the IL-6 cytokine family have been shown to synergistically drive astroglial differentiation in conjunction with BMPs, STAT3, and SMAD1 converging upon CBP/p300, respectively.10 We extend our previous observations5 and demonstrate scaleable generation of highly enriched astrocytes from HESCs through BMP4 and LIF application to late neural precursors. Accurate glial–neuronal disease modelling requires prior demonstration of function. While this study has been in preparation, Krencik et al.4 described the use of LIF/CNTF-mediated astrocyte differentiation from HESC-derived neural aggregates over 180 days, observing expression of astrocyte-specific markers including GFAP/S100β and glutamate uptake among other characterisation. We demonstrate the use of co-operative BMP4/LIF signalling for similar levels of astrocyte enrichment over a considerably shorter time course. The astrocytes generated in this study express a range of astrocyte markers; GFAP, AQP4, S100β, and EAAT1 and also crucially demonstrate functional glutamate uptake capacity.
There is growing evidence that astrocyte behaviour is dynamic, potentially either beneficial or detrimental for neuronal survival. Accumulating in-vivo evidence highlights a powerful neuroprotective effect of astrocytes.29, 30 We have shown previously in a rodent transgenic model of human P301S τ-mediated neurodegeneration, that age-related loss of neurons is reversed by astroglial transplantation.29 Other groups have shown similarly that astrocyte transplantation in a rodent transgenic model of ALS expressing human SOD1*G37R attenuated motor neuron loss and motor decline.30 Here, we demonstrate that HESC-derived astrocytes were able to protect neurons from oxidative injury by soluble factors, through a mechanism independent of de novo glutathione synthesis. Astrocytic supply of glutathione to neurons has been implicated as a key mediator of neuronal antioxidant defence;18, 31 baseline secretion of glutathione remains a potential neuroprotective mechanism as the duration of BSO treatment blocked de novo glutathione synthesis and did not fully deplete astrocytes of intracellular glutathione. Other soluble factor-mediated neuroprotective mechanisms include secretion of neurotrophic factors such as GDNF and IGF-1.29, 32
Oxidative stress is particularly prevalent in neurodegenerative injury11 and, as astrocytes are central to antioxidant defence in the CNS,33 modulation of astrocyte antioxidant abilities has been considered a potential therapeutic target in altering the clinical course of such disorders.12, 34 To this end, activators of Nrf2 have received considerable attention.12, 15, 17, 19, 35 Nrf2 modulation in astrocytes has a key role, abating neurological injury and disease progression in mouse models of ALS and Parkinson's disease in a glutathione-dependent manner.17, 19 We recently identified activation of astrocytic Nrf2 as a contributor to endogenous neuroprotective responses in the context of ischaemic preconditioning.15 The synthetic oleanane class of triterpenoids, which includes CDDO analogues, presents a promising drug treatment in multiple pathologies, due to their ability to modulate the activity of Nrf2.25 This study demonstrates that CDDOTFEA is capable of inducing strong neuroprotective responses in human astrocytes via a glutathione-dependent mechanism. Strikingly, human neurons derived from the same HESC line were completely incapable of responding to CDDOTFEA either in terms of activation of Nrf2 target genes, or by eliciting a neuroprotective effect. The reason behind this is unclear but may be due to low levels of NFE2L2 mRNA (which encodes Nrf2) in neurons (Figure 3a). However, it is consistent with the observation from mixed rodent cultures that astrocytes represent the major locus for Nrf2 activation, induced both by small molecules and by oxidative stress.15, 18
Non-cell-autonomous neuroprotection, as shown in our study, has widespread and fundamental implications for human-based drug discovery and screening of novel neuroprotective agents. Specifically, our findings suggest that screens focussing solely on the direct effect of compounds on isolated neurons will overlook potentially important neuroprotective activities that act via astrocytes or other non-neuronal cells. To conclude, these studies provide important proof-of-concept that human stem cell derived neural derivatives can be used effectively to model neuron–astrocyte interactions and investigate their mechanism. In the endeavour for clinically translatable interventions that may abate the course of neurodegenerative disorders, a key step in translation is the development of human in-vitro platforms, for both disease modelling and drug screening. HESC-based platforms overcome the issues of species-specific drug responses and transcriptional changes, and provide an important benchmark platform for studies using patient-specific pluripotent lines and clinical translation.
Materials and Methods
ES culture and neuralisation
The HESC lines H9 and HUES9 (Harvard University, Cambridge, MA, USA) were cultured and passaged regularly on a feeder layer of irradiated mouse embryonic fibroblasts, and neuralised by established protocols;21 In order to derive adherent hNPCs, HESC-derived neurospheres were dissociated and underwent selective propagation as an adherent monolayer on gelatin, in chemically defined medium36 supplemented with 20 ng/ml human FGF-2, heparin 5 μg/ml and 20 ng/ml human EGF, with 2-day complete medium changes. hNPCs were propagated for 2 months before astroglial differentiation. For astroglial differentiation, coverslips were coated with Matrigel (BD Biosciences, Oxford, UK) for 1 h, diluted 1 : 30 in Dulbecco's Modified Eagle's Medium (DMEM). Confluent hNPCs were enzymatically dissociated with Accutase and plated in basic plating medium (BPM) composed of 4.5 g/l glucose DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 2% B27 supplement minus antioxidants (Invitrogen) and 1% penicillin-streptomycin. BPM was modified for each plating condition with BMP2 and BMP4 (R&D, Abingdon, UK) at final concentrations of 10 ng/ml, and LIF (Sigma, St. Louis, MO, USA) at final concentrations of 20 ng/ml. Differentiating astrocytes received full media changes with factor-supplemented BPM every 2 days. Neurons underwent differentiation for 10 days in Neurobasal medium, supplemented with 2% B27 supplement minus antioxidants, 1% Glutamax (Gibco, Carlsbad, CA, USA), 1% non-essential amino acids, DAPT37 and 10 ng/ml BDNF.
Mouse mixed and astrocyte-free neuronal culture experiments
Cortical mouse mixed cultures of neurons and astrocytes were prepared as described38 from E17.5 Nrf2+/+ and Nrf2−/− mice with Neurobasal growth medium supplemented with B27 (Invitrogen). Typically, these cultures are ∼90% NeuN-positive neurons and 10% GFAP-positive astrocytes.39 To obtain astrocyte-free neuronal preparations (>98% NeuN+ neurons and <0.2% GFAP+ astrocytes15), cultures were treated with antimitotic cytosine-arabinoside immediately post-plating. Experiments were carried out on cultured neurons following a period of 10 days during which cortical neurons develop a network of processes, express functional NMDA-type and AMPA/kainate-type glutamate receptors, and form synaptic contacts. CDDOTFEA-induced neuroprotection against oxidative insults was studied as described previously for 3H-1,2-dithiole-3-thione-induced protection.35 Before the start of experiments, cultures were placed in minimal medium in the presence or absence of CDDOTFEA (100 nM), 16 h before an oxidative insult was applied in the form of H2O2 (100 μM, stabilised solution; Sigma). Cultures were fixed 24 h later and analysed after DAPI staining; cell death was quantified by counting (blind) the number of apoptotic nuclei as a percentage of the total. Approximately 1500 cells were counted per treatment, across four independent experiments. Morphologically, H2O2-treated neurons show typical signs of apoptotic-like cell death (shrunken cell body and large round chromatin clumps).
Gene expression analysis
Total RNA was isolated via the Absolutely RNA miniprep kit, cDNA synthesis performed via the AffinityScript cDNA (Stratagene, Stockport, UK) synthesis kit using oligo-dT and random hexamer primers, and qRT-PCR performed using Brilliant SYBR Green master mix (Stratagene), following the manufacturer's instructions. RT-PCRs were run over 28 cycles, and PCR products were separated on a 1.6% agarose in TBE gel, with GelRed for DNA visualisation. Primer sequences are listed in Supplementary Table S1.
Immunohistochemistry
Cells plated on glass coverslips were fixed with 4% paraformaldehyde for 20 min at room temperature and washed three times with PBS. Samples were next blocked for 1 h at room temperature with 0.2% Triton/PBS/5% goat serum and then incubated overnight with primary antibody in 0.2% Triton/PBS/2% goat serum at 4°C. After three washes in PBS, secondary antibody (Alexa Fluor, 1 : 1000) in PBS/DAPI (1 : 4000) was next applied for 1 h at room temperature. Primary antibodies used included Nestin (1 : 300; Chemicon, Bedford, MA, USA), 3CB2 (1 : 5; Developmental Studies Hybridoma Bank (DSHB), Iowa City, IA, USA), GFAP (1 : 300; Dako; 1 : 500; Abcam, Cambridge, UK), S100β (1 : 300; Dako, Ely, UK), AQP4 (1 : 50; Santa Cruz, Santa Cruz, CA, USA), EAAT1 (1 : 200; Novus Biologicals, Cambridge, UK), TuJ1 (1 : 500; Sigma-Aldrich) and activated caspase-3 (1 : 50; Chemicon).
Glutathione determination
Intracellular glutathione content was measured by the MCB (Sigma) assay. MCB is non-fluorescent in its native state and fluorescent upon conjugation with thiol-containing molecules including glutathione. While the MCB assay does not exclude other thiol-containing molecules in addition to glutathione, quantification of fluorescence has been used successfully to quantify GSH content.27 Cells were incubated in minimal medium containing 50 μM MCB for 45 min. Medium was removed and the cells were lysed; cell membranes were pelleted by centrifugation at (15 000 r.p.m., 5 min), and the fluorescence of the supernatant read by plate reader (FLUOstar OPTIMA, BMG Labtech, Aylesbury, UK) (excitation 405 nm/emission 520 nm). Fluorescence values obtained were adjusted for background signal and normalised to protein concentration, determined by BCA absorbance assay using bovine serum albumin standards.
Glutamate uptake
Plated astrocytes were washed twice in Tissue Buffer (5 mM Tris, 320 mM sucrose, pH 7.4) and then exposed to 10 μM 3H-glutamate in either Na+-containing Krebs buffer (120 mM NaCl, 25 mM NaHCO3, 5 mM KCl, 2 mM CaCl2, 1 mM KH2PO4, 1 mM MgSO4, 10% glucose) or Na+-free Krebs (120 mM choline-Cl and 25 mM Tris-HCl, pH 7.4 substituted for NaCl and NaHCO3, respectively) for 10 min at 37°C. Uptake was stopped by placing the astrocytes back on ice and washing them twice with Wash Buffer (5 mM Tris/160 mM NaCl pH 7.4). Cells were lysed with 0.1 M NaOH and radioactivity measured using a scintillation counter. Na+-dependent uptake was determined by subtracting Na+-free counts from total counts in the presence of Na+.
GCL enzyme kinetic assay
Reaction buffer and lysis buffer were prepared as described previously.40 ATP and cysteine hydrochloride were prepared fresh. Cells were lysed in 350 μl of lysis buffer, and cell debris pelleted (13 000 r.p.m., 10 min). In all, 50 μl of cell lysate was placed at 37°C for each of five time points, and 50 μl of reaction buffer and 40 mM ATP added. After 5 min equilibration, 50 μl 20 mM cysteine hydrochloride was added, for 30, 20, 10, and 5 min, and at time 0. The reaction was terminated by the addition of 50 μl metaphosphoric acid, and placement on ice. Precipitated proteins were pelleted (2500 r.p.m., 5 min), and glutamyl-cysteine quantified by NDA fluorescence. In all, 20 μl of reaction mixture was combined with 180 μl of NDA reaction mixture, comprising 140 μl Trizma-base 50 mM, 20 μl 1 M NaOH, 20 μl NDA 10 mM, and fluorescence determined by plate reader (excitation 485 nm/emission 520 nm).
Conditioned medium generation
Astrocytes were treated with minimal medium, consisting of Salt–Glucose–Glycine39 supplemented with 10% MEM (Invitrogen), in the presence and absence of drugs for 6 h, after which the drugs were washed out and fresh minimal medium replaced. Drugs included 1[2-Cyano-3,12-dioxool-eana-1,9(11)-dien-28-oyl] trifluoroethylamide (CDDOTFEA; 250 nM), BSO (200 μM), and bischloronitrosurea (BCNU; 50 μM). After 24 h, ACM was removed, cell debris pelleted (4°C, 3400 r.p.m. for 10 min), and half-supplemented with fresh minimal medium before the addition of H2O2 and neuronal viability determined.
Neuronal injury and cell viability assays
Neurons were preconditioned in minimal medium for 12 h, before addition of control or astrocyte-conditioned minimal medium supplemented with H2O2. After peroxidative challenge, cell viability was quantified using CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA), following the manufacturer's instructions. In brief, assay reagents were reconstituted according to instructions, and an equal volume added to cell medium. After 10 min, cell lysate was removed and luminescence quantified by plate reader.
Data analysis
All experiments were performed with at least three biological repeats; within each, three technical repeats were generated. Experiments were repeated across two independent HESC lines. qPCR data were normalised to GAPDH. For quantitative IHC, five random views were taken per coverslip, and cell counts were performed manually, assessing staining specificity and morphology. Data are presented as mean±standard error of the mean; statistical analyses were performed in Microsoft Excel and GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA, USA) by univariate and two-way ANOVA with Newman–Keuls post-test and the Students’ t-test. A P-value of 0.05 was considered statistically significant.
Abbreviations
- ACM:
-
astrocyte-conditioned medium
- BCNU:
-
bischloronitrosourea
- BSO:
-
buthionine sulfoximine
- CDDOTFEA:
-
1[2-Cyano-3,12-dioxool-eana-1,9(11)-dien-28-oyl] trifluoroethylamide
- GCL:
-
glutamate-cysteine ligase
- HESC:
-
human embryonic stem cell
- hNPC:
-
human neural precursor cell
References
Di Giorgio F, Boulting G, Bobrowicz S, Eggan K . Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 2008; 3: 637–648.
Marchetto M, Muotri A, Mu Y, Smith A, Cezar G, Gage F . Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell 2008; 3: 649–657.
Zhang S, Wernig M, Duncan I, Brustle O, Thomson J . In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol 2001; 19: 1129–1133.
Krencik R, Weick JP, Liu Y, Zhang ZJ, Zhang SC . Specification of transplantable astroglial subtypes from human pluripotent stem cells. Nat Biotechnol 2011; 29: 528–534.
Joannides A, Webber D, Raineteau O, Kelly C, Irvine K, Watts C et al. Environmental signals regulate lineage choice and temporal maturation of neural stem cells from human embryonic stem cells. Brain 2007; 130 (Part 5): 1263–1275.
Molne M, Studer L, Tabar V, Ting Y, Eiden M, McKay R . Early cortical precursors do not undergo LIF-mediated astrocytic differentiation. J Neurosci Res 2000; 59: 301–311.
Naka H, Nakamura S, Shimazaki T, Okano H . Requirement for COUP-TFI and II in the temporal specification of neural stem cells in CNS development. Nat Neurosci 2008; 11: 1014–1023.
Barnabe-Heider F, Wasylnka JA, Fernandes KJ, Porsche C, Sendtner M, Kaplan DR et al. Evidence that embryonic neurons regulate the onset of cortical gliogenesis via cardiotrophin-1. Neuron 2005; 48: 253–265.
Yanagisawa M, Nakashima K, Takizawa T, Ochiai W, Arakawa H, Taga T . Signaling crosstalk underlying synergistic induction of astrocyte differentiation by BMPs and IL-6 family of cytokines. FEBS Lett 2001; 489: 139–143.
Nakashima K, Yanagisawa M, Arakawa H, Kimura N, Hisatsune T, Kawabata M et al. Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science 1999; 284: 479–482.
Andersen J . Oxidative stress in neurodegeneration: cause or consequence? Nat Med 2004; 10 (Suppl): S18–S25.
Vargas MR, Johnson JA . The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev Mol Med 2009; 11: e17.
Kensler TW, Wakabayashi N, Biswal S . Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol 2007; 47: 89–116.
McMahon M, Itoh K, Yamamoto M, Hayes JD . Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem 2003; 278: 21592–21600.
Bell KF, Al-Mubarak B, Fowler JH, Baxter PS, Gupta K, Tsujita T et al. Mild oxidative stress activates Nrf2 in astrocytes, which contributes to neuroprotective ischemic preconditioning. Proc Natl Acad Sci USA 2011; 108: E1–E2; author reply E3--E4.
Haskew-Layton RE, Payappilly JB, Smirnova NA, Ma TC, Chan KK, Murphy TH et al. Controlled enzymatic production of astrocytic hydrogen peroxide protects neurons from oxidative stress via an Nrf2-independent pathway. Proc Natl Acad Sci USA 2010; 107: 17385–17390.
Vargas M, Johnson D, Sirkis D, Messing A, Johnson J . Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci 2008; 28: 13574–13581.
Shih A, Johnson D, Wong G, Kraft A, Jiang L, Erb H et al. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci 2003; 23: 3394–3406.
Chen P, Vargas M, Pani A, Smeyne R, Johnson D, Kan Y et al. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson's disease: critical role for the astrocyte. Proc Natl Acad Sci USA 2009; 106: 2933–2938.
Oberheim NA, Takano T, Han X, He W, Lin JH, Wang F et al. Uniquely hominid features of adult human astrocytes. J Neurosci 2009; 29: 3276–3287.
Joannides A, Fiore-Heriche C, Battersby A, Athauda-Arachchi P, Bouhon I, Williams L et al. A scaleable and defined system for generating neural stem cells from human embryonic stem cells. Stem Cells 2007; 25: 731–737.
Koch P, Opitz T, Steinbeck J, Ladewig J, Brustle O . A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc Natl Acad Sci USA 2009; 106: 3225–3230.
Rothstein J, Martin L, Levey A, Dykes-Hoberg M, Jin L, Wu D et al. Localization of neuronal and glial glutamate transporters. Neuron 1994; 13: 713–725.
Mahesh VB, Dhandapani KM, Brann DW . Role of astrocytes in reproduction and neuroprotection. Mol Cell Endocrinol 2006; 246: 1–9.
Stack C, Ho D, Wille E, Calingasan N, Williams C, Liby K et al. Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington's disease. Free Radic Biol Med 2010; 49: 147–158.
Yates MS, Tauchi M, Katsuoka F, Flanders KC, Liby KT, Honda T et al. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther 2007; 6: 154–162.
Barhoumi R, Bailey R, Burghardt R . Kinetic analysis of glutathione in anchored cells with monochlorobimane. Cytometry 1995; 19: 226–234.
Barres BA . The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 2008; 60: 430–440.
Hampton D, Webber D, Bilican B, Goedert M, Spillantini M, Chandran S . Cell-mediated neuroprotection in a mouse model of human tauopathy. J Neurosci 2010; 30: 9973–9983.
Lepore A, Rauck B, Dejea C, Pardo A, Rao M, Rothstein J et al. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat Neurosci 2008; 11: 1294–1301.
Bolanos JP, Heales SJ, Peuchen S, Barker JE, Land JM, Clark JB . Nitric oxide-mediated mitochondrial damage: a potential neuroprotective role for glutathione. Free Radic Biol Med 1996; 21: 995–1001.
Garcia-Estrada J, Garcia-Segura L, Torres-Aleman I . Expression of insulin-like growth factor I by astrocytes in response to injury. Brain Res 1992; 592: 343–347.
Dringen R, Pfeiffer B, Hamprecht B . Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci 1999; 19: 562–569.
Gegg ME, Beltran B, Salas-Pino S, Bolanos JP, Clark JB, Moncada S et al. Differential effect of nitric oxide on glutathione metabolism and mitochondrial function in astrocytes and neurones: implications for neuroprotection/neurodegeneration? J Neurochem 2003; 86: 228–237.
Soriano FX, Leveille F, Papadia S, Higgins LG, Varley J, Baxter P et al. Induction of sulfiredoxin expression and reduction of peroxiredoxin hyperoxidation by the neuroprotective Nrf2 activator 3H-1,2-dithiole-3-thione. J Neurochem 2008; 107: 533–543.
Vallier L, Pedersen R . Differentiation of human embryonic stem cells in adherent and in chemically defined culture conditions. Curr Protoc Stem Cell Biol 2008; Chapter 1: Unit 1D.4.1--1D.4.7.
Borghese L, Dolezalova D, Opitz T, Haupt S, Leinhaas A, Steinfarz B et al. Inhibition of notch signaling in human embryonic stem cell-derived neural stem cells delays G1/S phase transition and accelerates neuronal differentiation in vitro and in vivo. Stem Cells 2010; 28: 955–964.
Leveille F, Papadia S, Fricker M, Bell KF, Soriano FX, Martel MA et al. Suppression of the intrinsic apoptosis pathway by synaptic activity. J Neurosci 2010; 30: 2623–2635.
Papadia S, Soriano FX, Leveille F, Martel MA, Dakin KA, Hansen HH et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci 2008; 11: 476–487.
White CC, Viernes H, Krejsa CM, Botta D, Kavanagh TJ . Fluorescence-based microtiter plate assay for glutamate-cysteine ligase activity. Anal Biochem 2003; 318: 175–180.
Acknowledgements
We thank Professor Michael Sporn for the gift of CDDOTFEA, Professor Jeff Johnson for comments on the manuscript, and Professor Masayuki Yamamoto of the University of Tsukuba (now University of Tohoku) for kindly providing Nrf2−/− mice. TT and JDH wish to thank the Association for International Cancer Research for their support (Grant 07-0074). This work was funded by the Evelyn Trust, Motor Neuron Disease Association, Medical Research Council, and the Wellcome Trust.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Deshmukh
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Gupta, K., Patani, R., Baxter, P. et al. Human embryonic stem cell derived astrocytes mediate non-cell-autonomous neuroprotection through endogenous and drug-induced mechanisms. Cell Death Differ 19, 779–787 (2012). https://doi.org/10.1038/cdd.2011.154
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2011.154
Keywords
This article is cited by
-
Astrocyte-oligodendrocyte interaction regulates central nervous system regeneration
Nature Communications (2023)
-
Targeted de-repression of neuronal Nrf2 inhibits α-synuclein accumulation
Cell Death & Disease (2021)
-
Glial cells in Parkinson´s disease: protective or deleterious?
Cellular and Molecular Life Sciences (2020)
-
Complement 3+-astrocytes are highly abundant in prion diseases, but their abolishment led to an accelerated disease course and early dysregulation of microglia
Acta Neuropathologica Communications (2019)
-
Prevention of neuronal apoptosis by astrocytes through thiol-mediated stress response modulation and accelerated recovery from proteotoxic stress
Cell Death & Differentiation (2018)
