
Abstract
Apoptosis is regulated by changes in the subcellular distribution of pro- and anti-apoptotic proteins, among which are nuclear proteins such as histone H1 (H1) and nucleophosmin (NPM). These proteins were reported to translocate to the cytosol and mitochondria, and to facilitate apoptosis in response to apoptotic stressors. The significance of this stress-induced, nuclear protein redistribution and its exact molecular mechanism are poorly understood. We show here that in mouse embryonic fibroblasts (MEFs), different apoptotic stimuli induce H1, NPM and nucleolin, but not KAP-1 nuclear/cytoplasmic redistribution, which precedes the appearance of apoptotic features. Using MEFs deficient in Bax/Bak, Apaf-1 or caspase-9, as well as caspase inhibitors, we show that this redistribution requires Bax and Bak, but neither the apoptosome nor caspases. Furthermore, the BH3 mimetic ABT-737, which acts through Bax/Bak, also stimulates nuclear protein redistribution in a Bax/Bak-dependent manner. Re-expression of Bax or Bak in Bax/Bak-deficient MEFs restores nuclear redistribution during apoptosis. This is not accompanied by Bax or Bak N-terminus exposure and is not inhibited by Bcl-xL overexpression. These results identify, for the first time, a function of Bax/Bak that is insensitive to inhibition by Bcl-xL and most likely unrelated to their canonical, pore-forming activity on mitochondria.
Similar content being viewed by others


Main
The Bcl-2 protein family consists of anti- and pro-apoptotic members. The anti-apoptotic proteins include Bcl-2 and Bcl-xL, whereas the pro-apoptotic members include the multi-domain proteins Bax and Bak, and the BH3-only proteins.1, 2 Experiments using cells derived from mice lacking both Bax and Bak (Bax/Bak double knockout (Bax/Bak DKO)) showed that Bax and Bak are key regulators of the mitochondria-mediated apoptotic pathway.3, 4, 5 In healthy cells, Bax exists as an inactive monomer in the cytoplasm, whereas Bak is already inserted in the mitochondrial outer membrane (MOM). During apoptosis, Bax translocates to the MOM and Bak is relieved from inhibition by unknown mechanisms. Subsequently, both Bax and Bak undergo conformational changes, consequently exposing their N-terminal (NT) regions and forming homo- and hetero-oligomers.6 The Bax/Bak oligomers perforate the MOM, consequently releasing apoptogenic factors such as cytochrome c. The binding of cytochrome c to Apaf-1 generates the Apaf-1/caspase-9 apoptosome and subsequently activates effector caspases-3 and -7.5
Cells often use the translocation of apoptotic proteins from one cellular compartment to another to regulate apoptosis. Apart from Bax and cytochrome c, other examples of such proteins include the nuclear proteins p53,7, 8 Nur77,9 caspase-2,10 nucleophosmin (NPM),11, 12 and histone H1.2.13, 14 During apoptosis, all these proteins migrate from the nucleus to the cytosol and/or to mitochondria, where they participate in crucial steps of apoptosis. The mechanisms underlying nuclear/cytoplasmic redistribution and particular apoptotic pathways involved remain to be elucidated.
The aim of this study was to identify the signaling pathway that promotes nuclear protein redistribution during apoptosis. To this end, we used MEFs as a cellular model system and focused on three different nuclear proteins: NPM, histone H1 (H1) and nucleolin. NPM is a multifunctional nucleolar phosphoprotein controlling vital cell processes such as ribosome biogenesis, DNA repair and RNA transcription.11 It was suggested to regulate Bax translocation and activation by interacting with a conformationally altered Bax.15, 16 H1 participates in the formation of high-order chromatin structures, and thereby inhibits RNA transcription.17 A particular isoform of H1, H1.2, was found to co-localize with Bak and to contribute to cytochrome c release and apoptosis in a Bak-dependent manner.13, 14 Nucleolin is a nucleolar protein involved in chromatin remodeling, DNA recombination and replication, RNA transcription, rRNA processing and mRNA stabilization.18, 19 Under stress conditions such as herpes simplex virus type 1 infection20 or DNA damage,21 nucleolin redistributes from nucleoli to the nucleoplasm and to the cytosol.
Our results show that in MEFs apoptotic stimuli induce the redistribution of all three proteins before cytochrome c release, caspase activation and morphological signs of apoptosis. Strikingly, the redistribution process does not involve classical NT conformational changes of Bax or Bak and depends on a new function of the two proapoptotic proteins that cannot be inhibited by Bcl-xL overexpression.
Results
Stress-induced redistribution of NPM, H1 and nucleolin, but not of KAP-1, occurs early after inducing apoptosis, independently of apoptosome and caspases
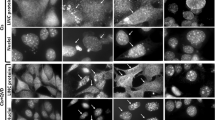
First, we wanted to confirm that various apoptotic stimuli change the subcellular distribution of nuclear proteins NPM, H1 and nucleolin, a process that, in this study, is referred to as ‘redistribution’. Thus, we treated wild-type (WT) MEFs with 25 μM cisplatin, 1 μM camptothecin, 1 μM doxorubicin or 100 nM staurosporine for different times and monitored the redistribution by immunofluorescence analysis. As expected in healthy MEFs, all three proteins mainly resided in the nucleus. NPM and nucleolin were confined to nucleoli (Figure 1a and b), whereas H1 was present throughout the nucleus (Figure 1c). In response to apoptotic stimuli, MEFs underwent time-dependent apoptosis, as determined by annexin-V/PI FACS analysis (Supplementary Figure S1a). Concomitantly with the death process, the nuclear distribution of all three proteins changed dramatically, but each protein showed a distinct behavior. NPM was evenly distributed in the cytoplasm, and this sometimes correlated with a reduced expression in the nuclei and nucleoli (Figure 1a). Nucleolin (Figure 1b) also appeared in the cytoplasm but was considerably less than NPM. Higher magnifications revealed a granular immunofluorescence pattern of cytosolic nucleolin, distributed throughout the cytoplasm without being confined to a particular subcellular compartment such as mitochondria (data not shown). Sometimes nucleolin redistribution was hardly detected, both in the nuclei and in the cytosol. This was probably because of partial nucleolin degradation and not cell destruction because Hoechst 33258 staining still revealed intact nuclei.22 Finally, the nuclear staining of H1 staining was markedly reduced, and a low amount of punctuated, extranuclear H1 immunofluorescence was observed in response to apoptotic stressors (Figure 1c and d). This punctuated pattern is reminiscent of that previously reported for cytosolic H1 in stress-induced thymocytes and MEFs,13, and was partially associated with mitochondria (data not shown). The quantification of NPM, nucleolin and H1 redistribution by individual cell counting revealed that although this process also occurred in untreated MEFs at low frequency (12±2, 1±0.2 and 3±2% for NPM, H1, and nucleolin, respectively), it drastically increased for all three proteins in cells exposed to cisplatin, camptothecin, doxorubicin or staurosporine (Figure 1e). Importantly, the cisplatin-induced redistribution process could be confirmed by monitoring direct GFP fluorescence in WT MEFs expressing GFP–nucleolin. Although in untreated MEFs, GFP–nucleolin was confined to the nucleus and only few cells (2%) showed a cytosolic GFP in response to cisplatin, 70% of the GFP–nucleolin expressing cells showed a cytosolic GFP (Figure 1f and g). Assessment of the stress-induced redistribution of an additional nuclear protein, KAP-1, revealed that KAP-1 did not change its localization in response to cisplatin or camptothecin (Supplementary information and Supplementary Figure S2), therefore suggesting that stress induces the redistribution for some, but not all, nuclear proteins.


(a–e) Redistribution of NPM, nucleolin and H1 in response to apoptotic stimuli. WT MEFs were untreated (Con) or treated for 24 h with 25 μM cisplatin (Cis), 1 μM camptothecin (Camp) or 1 μM doxorubicin (Doxo), or treated with 100 nM staurosporine (STS) for 17 h, followed by staining with anti-NPM (a), anti-nucleolin (b) or anti-H1 (c) antibodies, and with the Hoechst 33258 dye (for staining the nuclei), and thereafter visualized by fluorescence microscopy. The images of each treatment (upper and lower panels) represent the same field visualized separately for detecting antibody staining and Hoechst-stained nuclei. The results presented are from a representative experiment (one of at least four independent experiments). Arrows indicate the cells exhibiting the redistribution of a nuclear protein and their nuclei. Higher magnification of a representative area (denoted by a box in the staurosporine image) illustrating extranuclear-punctuated H1 staining is shown in (d). Quantification of NPM, H1 and nucleolin redistribution is shown in (e). The number of cells exhibiting redistribution of NPM, H1 and nucleolin was determined microscopically. The results presented are expressed as the percentage of cells exhibiting redistribution of each nuclear protein from all the cells counted (at least 200 cells) in each treatment. The values are represented as means±S.E.M. (bars; n=3). Bars (a–c) 20 μm; (d) 5 μm. *P<0.05; **P<0.01, which is significantly higher than that of the corresponding untreated cells (Con) (one-tailed Student's t-test). (f and g) The effect of cisplatin on GFP–nucleolin subcellular distribution. MEFs stably expressing GFP–nucleolin were untreated (Con) or treated for 24 h with 25 μM cisplatin (Cis). After treatment, the nuclei were stained with Hoechst 33258 as described in Materials and Methods and thereafter, GFP–nucleolin and nuclei were visualized by fluorescence microscopy. The images (f) for each treatment (left and right panels) represent the same field visualized separately for detecting GFP–nucleolin and Hoechst-stained nuclei. The results presented are from a representative experiment (one of at least three independent experiments). Quantification of GFP–nucleolin redistribution is shown in (g). The number of cells exhibiting redistribution of GFP–nucleolin was determined microscopically. The results presented are expressed as the percentage of cells exhibiting GFP–nucleolin redistribution from all the cells counted (at least 200 cells) in each treatment. The values shown are the means±S.E.M. (bars; n=3). Bars, 20 μm; **P<0.01, which is significantly higher than that of untreated cells (Con) (one-tailed Student's t-test). camp, camptothecin; cis, cisplatin; Con, untreated; doxo, doxorubicin; GFP, green fluorescent protein; H1, histone 1; MEFs, mouse embryonic fibroblasts; NPM, nucleophosmin; STS, staurosporine; WT, wild type
Next, we carried out a time-course analysis of nuclear protein redistribution in cisplatin- and camptothecin-treated WT MEFs (Figure 2a). In response to cisplatin (upper panel, Figure 2a), the redistribution of NPM, H1 and nucleolin already started at 2 h, plateaued at 6–9 h and then further increased to maximal values by 24 h. In contrast, almost no KAP-1 redistribution (∼1%) was observed at 2–9 h and only 11% was observed at 24 h. It is noteworthy that at 6–9 h of cisplatin treatment, when about 30% of cells exhibited nuclear protein redistribution (Figure 2a), very few cells (1–3%) exhibited apoptotic features, such as Bax or Bak NT exposure, caspase-3 activation, cytochrome c release, apoptotic nuclei (Figure 2b, upper panel) or His–GFP–annexin-V exposure (Supplementary Figure S1a). Similar results were obtained when WT MEFs were treated with camptothecin (Figure 2a and b, middle panel). To exclude the possibility that these results were biased toward the WT MEF cell clone used, we examined nuclear protein redistribution and apoptotic features in a WT MEF cell line isolated independently (termed WT1). As shown in Figure 2a (lower panel), cisplatin induced a time-dependent nuclear protein redistribution in WT1 MEFs, resembling that in WT MEFs, although with moderate differences (e.g., the extent of redistribution of nucleolin and NPM was higher in WT1 MEFs) and also preceded the appearance of apoptotic events (Figure 2b, lower panel). Collectively, these results suggest that the redistribution of NPM, H1 and nucleolin represents an early stress response that occurred before Bax/Bak activation, cytochrome c release and caspase-3 activation.

Time-course analysis of nuclear protein redistribution (a) and apoptotic events (b) in WT and WT1 MEFs. WT and WT1 MEFs were treated with 25 μM cisplatin, or WT MEFs were treated with 1 μM camptothecin for 2, 4, 6, 9 and 24 h and then stained with anti-NPM, anti-H1, anti-nucleolin, anti-KAP-1, anti-Bax NT, anti-Bak NT, anti-cytochrome c or anti-active caspase 3 (Active Cas-3) antibodies, together with Hoechst 33258 (to detect apoptotic (Ap) nuclei). The number of cells exhibiting nuclear protein redistribution or each of the indicated apoptotic parameters was determined microscopically. Apoptotic nuclei were defined as nuclei that exhibited condensed or fragmented morphology. Cytochrome c release (Cyt c) was defined as cells exhibiting a diffused staining pattern. The results presented are expressed as the percentage of cells exhibiting a redistribution of each nuclear protein (a) or apoptotic events (b) from all the cells (at least 200 cells) counted, at each time point. The values are represented as means±S.E.M. (bars; n=3). Cyt c, cytochrome c; H1, histone 1; KAP1, KRAB-associated protein 1; MEFs, mouse embryonic fibroblasts; NPM, nucleophosmin; NT, N-terminal; WT, wild type
Next, we examined the role of the apoptosome and caspases in stress-induced nuclear protein redistribution. First, we treated Apaf-1−/− MEFs with cisplatin, camptothecin, doxorubicin or staurosporine, as described above. As previously reported23 and shown in Supplementary Figure S1b, Apaf-1−/− MEFs were found to be resistant to apoptosis induced by these drugs. However, despite this resistance, the stress-induced redistribution of NPM, nucleolin and H1 was not affected by Apaf-1 deficiency (Figure 3a–c). Quantification of nuclear protein redistribution in WT (Figure 1e) and Apaf-1−/− MEFs (Figure 3d) revealed that the percentages of cells showing this effect after 24 h of drug treatment were similar in both genotypes for all three nuclear proteins, although basal levels (untreated) of redistribution were elevated in Apaf-1−/− cells, especially for NPM. To support our data, we also assessed the process in caspase-9−/− MEFs. Again, although cell death was inhibited in caspase-9−/− cells (data not shown), redistribution of nucleolin in response to cisplatin or camptothecin occurred in a manner similar (Supplementary Figure S3) to that in Apaf-1−/− (Figure 3d) and WT MEFs (Figure 1e). Similarly, redistribution of nucleolin also occurred when WT cells were treated with the general caspase inhibitor, Boc-Asp(OMe)-FMK (Boc) (Supplementary information and Supplementary Figure S4), thus excluding the role of other caspases in the redistribution effect. Collectively, our results indicate that stress-induced redistribution of nuclear proteins NPM, nucleolin and H1 occurs through a mechanism independent of the apoptosome and caspases.

Apaf-1 is not needed for stress-induced NPM, nucleolin and H1 redistribution. Apaf-1−/− MEFs were treated, stained and visualized as described in Figure 1. The results presented for NPM (a), nucleolin (b) and H1 (c) are from a representative experiment (one of at least four independent experiments). Arrows indicate cells exhibiting the redistribution of a nuclear protein and their nuclei. Quantification of NPM, H1 and nucleolin redistribution is shown in (d). The number of cells exhibiting a redistribution of NPM, H1 and nucleolin was determined and expressed as described in Figure 1e. The values are represented as means±S.E.M. (bars; n=3). *P<0.05; **P<0.01, which is significantly higher than that of the corresponding untreated cells (Con) (one-tailed Student's t-test). Bars, (a–c) 20 μm. H1, histone 1; MEFs, mouse embryonic fibroblasts; NPM, nucleophosmin
The results depicted in Figure 2 clearly show that nuclear protein redistribution precedes the appearance of apoptotic features (cytochrome c release, Bax/Bak NT exposure and fragmented nuclei) when a single-cell analysis was performed by immunostaining matrix-attached cells. This, however, underestimates the total number of apoptotic cells because these cells tend to detach from the matrix. To overcome this limitation, we examined the relationship between the nuclear protein redistribution effect and the appearance of apoptotic features in caspase-9−/− MEFs, known to be quite resistant to apoptosis. We chose these cells because their basal nuclear protein redistribution is lower than that in Apaf-1−/− MEFs. As expected, no major cell death occurred in caspase-9−/− MEFs after exposure to cisplatin for 9, 17, 24 and 48 h (data not shown). Moreover, cytochrome c release and Bax/Bak NT exposure were barely detected at 9 h (Figure 4b). However, substantial amounts of NPM, nucleolin and H1 were already redistributed at this time (31, 63 and 20%, respectively) and the redistribution gradually increased to 66, 100 and 76% at 48 h, respectively (Figure 4a). From 17 h onward, Bax/Bak NT exposure and cytochrome c release began to increase, but the cells remained attached to the plate (Figure 4b; data not shown). These results show that nuclear protein redistribution is not a consequence of cell destruction, but occurs as an early stress response that precedes Bax/Bak activation and cytochrome c release.

Time-course analysis of nuclear protein redistribution (a) and apoptotic events (b) in caspase-9−/− MEFs. Caspase-9−/− MEFs were treated with 25 μM cisplatin for 9, 17, 36, and 48 h and then stained with anti-NPM, anti-H1, anti-nucleolin, anti-Bax NT, anti-Bak NT or anti-cytochrome c. The results presented are expressed as described in Figure 2. The values are represented as means±S.E.M. (bars; n=3). H1, histone 1; MEFs, mouse embryonic fibroblasts; NPM, nucleophosmin; NT, N-terminal
Stress-induced redistribution of NPM, H1 and nucleolin requires Bax and Bak
As the redistribution of nuclear proteins preceded cytochrome c release, we wanted to determine whether it required the activation of Bax and Bak, an obligatory step for MOM perforation. MEFs deficient in both Bax and Bak were treated with cisplatin, camptothecin, doxorubicin or staurosporine, as described above. As reported,3, 4 Bax/Bak DKO MEFs were found to be highly resistant to apoptosis induced by these treatments (Supplementary Figure S1c). However, in contrast to WT and Apaf-1−/− MEFs (Figures 1 and 3), the redistribution of NPM, nucleolin and H1 was effectively blocked in drug-treated Bax/Bak DKO MEFs (Figure 5). Lesser inhibition was detected with H1 redistribution in staurosporine-treated Bax/Bak DKO MEFs, although this redistribution (30%; Figure 5d) was still much lower than that in WT (89%; Figure 1d) and Apaf-1−/− (97%; Figure 3d) MEFs. It is noteworthy that the lack of stress-induced nuclear protein redistribution in Bax/Bak DKO cells was not because of the unresponsiveness of these cells to stress stimuli because, for example, NPM was still redistributed from the nucleoli to the nucleoplasm in response to doxorubicin, although a further redistribution to the cytoplasm did not occur (Figure 5a). To confirm our findings, we transiently transfected GFP–nucleolin and GFP–NPM into WT and Bax/Bak DKO MEFs and found that, in contrast to WT cells, the redistribution of both proteins was impeded in the absence of Bax/Bak (Supplementary information and Supplementary Figure S5). These data indicate that Bax and Bak are required for the stress-induced nuclear protein redistribution effect.

(a-c) Bax and Bak are needed for stress-induced NPM, nucleolin and H1 redistribution. Bax/Bak DKO MEFs were treated, stained and visualized as described in Figure 1. The results presented are from a representative experiment (one of at least four experiments). Quantification of NPM, H1 and nucleolin redistribution is shown in (d). The number of cells exhibiting a redistribution of NPM, H1 and nucleolin was determined and expressed as described in Figure 1e. The values shown represent the means±S.E.M. (bars; n=3). *P<0.05, which is significantly higher than that of the corresponding untreated cells (Con) (one-tailed Student's t-test). Bars, (a–c)=20 μm. DKO, double knock out; H1, histone 1; MEFs, mouse embryonic fibroblasts; NPM, nucleophosmin
It is well known that Bax and Bak are redundant in mediating apoptosis.4 To determine whether this is also the case regarding their effects on nuclear protein redistribution, we examined this process in response to cisplatin in MEFs singly deficient for either Bax or Bak. As expected, Bax−/− and Bak−/− MEFs underwent cisplatin-induced apoptosis (data not shown). Moreover, in single knockout cells, the number of cells showing nucleolin redistribution after 24 h of drug treatment was similar to that of WT MEFs (Supplementary Figure S6). Thus, either Bax or Bak can similarly mediate stress-induced redistribution of nuclear proteins.
The stress-induced H1 or NPM redistribution effect is uncoupled from NT exposure of Bax and Bak
As the stress-induced redistribution of NPM, H1 and nucleolin was Bax/Bak dependent, it was important to determine whether this process is mediated through the classical proapoptotic function of Bax and Bak on mitochondria. An early step in the activation of Bax and Bak is the conformational change at their N-termini, which is crucial for their pore-forming activity on the MOM.6 This can be detected by immunofluorescence analysis using a conformation-specific antibody. Figure 6 shows a single-cell analysis of WT MEFs treated with cisplatin for 24 h and co-stained with anti-NPM or anti-H1 antibodies, and the conformation-specific anti-Bax or anti-Bak antibodies (Bax NT or Bak NT). Unexpectedly, whenever NPM and H1 were redistributed, the conformational change of Bax and Bak was found to be rare (Figure 6a). Quantitative analysis revealed that only about 10 and 20% of the cells showing redistribution of NPM and H1, respectively, were positive for Bax or Bak NT staining. To bolster our findings and to statistically evaluate the relationship between the appearance of Bax or Bak NT exposure and nuclear protein redistribution, we again used caspase-9−/− MEFs because these cells did not undergo apoptosis and thus more cells were available for the analysis (see Figure 4b). The percentage of cells that exhibited Bax or Bak NT exposure in the cell population showing H1 or NPM redistribution, as well as the percentage cells that exhibited H1 or NPM redistribution in the cell population showing Bax or Bak NT exposure, at 24, 36 and 48 h after cisplatin treatment is shown in Figure 6b. A statistical analysis by χ2 test (df=2) (for details, see Materials and Methods) revealed that at all times and for all parameters examined, the proportion of cells that exhibited both nuclear protein redistribution and NT exposure was lower than the values anticipated if the two events were independent (P⩽2.1 × 10−18). This suggests that the probability that nuclear protein redistribution (NPM or H1) occurs together with NT exposure (Bax or Bak) in the same cell is not random. Moreover, the appearance of one event lowers the probability of the other appearing. Therefore, the effect of Bax and Bak on the redistribution of nuclear proteins is uncoupled from NT conformational change.

The relationship between nuclear protein redistribution and Bax/Bak NT exposure. (a) WT MEFs were treated with 25 μM cisplatin and 100 μM Boc for 24 h, then double stained with anti-NPM or anti-H1 antibodies together with anti-Bax NT (left panel) or anti-Bak NT (right panel), and with Hoechst 33258, and visualized by fluorescence microscopy. The images represent the same field visualized separately for detecting antibody double staining (top and middle panel) and Hoechst-stained nuclei (lower panel). The results presented are from a representative experiment (one of at least four independent experiments). Arrowheads indicate cells and their nuclei that exhibit protein redistribution without Bax NT or Bak NT staining. Arrows indicate cells and their nuclei that exhibit Bax NT or Bak NT staining but not nuclear protein redistribution. Asterisks indicate a cell and its nucleus that exhibit nuclear protein redistribution and Bak NT staining (lower panel). Quantification of the number of cells exhibiting NPM or H1 redistribution, together with Bax or Bak NT exposure. Cells were treated and stained as described above. The results presented are expressed as the percentage of cells exhibiting Bax or Bak NT from those cells showing nuclear protein redistribution (of each NPM or H1) (n⩾100 cells). Values are represented as means±S.E.M. (bars; n=3). Bars, 20 μm. (b) Caspase-9−/− MEFs were treated as described in Figure 4. After 24, 36 and 48 h, the cells were double stained for anti-H1 or anti-NPM, together with anti-Bax NT or anti-Bak NT. The results are expressed as the percentage of cells exhibiting Bax or Bak NT exposure compared with those cells showing H1 or NPM redistribution (n=100) (Bax from H1 or Bak from H1; Bax from NPM or Bak from NPM) or the percentage of cells showing H1 or NPM redistribution compared with those showing Bax or Bak NT exposure (n=100) (H1 from Bax or H1 from Bak; NPM from Bax or NPM from Bak). The values are represented as means±S.E.M. (bars; n=3). MEFs, mouse embryonic fibroblasts; NPM, nucleophosmin; NT, N-terminal; WT, wild type
Ectopic expression of Bcl-xL does not inhibit stress-induced NPM, H1 and nucleolin redistribution
To further support the idea that Bax and Bak may mediate nuclear protein redistribution through a noncanonical function, we stably expressed the prosurvival protein, Bcl-xL, in the form of a tagged construct (FLAG–Bcl-xL) in WT MEFs (Bcl-xL cells). Overexpression of Bcl-xL is known to inhibit MOM perforation and all subsequent apoptotic events by interacting with activated Bax and Bak.2, 24 Indeed, although vector control (pcDNA3) MEFs exhibited 30% of apoptotic nuclei after 24 h of cisplatin treatment, only few such nuclei (3%) were detected in FLAG–Bcl-xL-expressing MEFs (Figures 7 and 8b). Moreover, none of the latter displayed anti-Bax NT exposure or cytochrome c release (Figure 8b, upper and middle panels, respectively). However, the redistributions of NPM, H1 and nucleolin were not affected by FLAG–Bcl-xL overexpression (Figure 7a). Quantitative analysis revealed that a similar number of vector control or FLAG–Bcl-xL-expressing cells exhibited nuclear protein redistribution after 24 h of cisplatin treatment (Figure 8a). Furthermore, as seen above in Apaf-1−/− MEFs (Figure 3d), the basal level of the redistribution of NPM was moderately increased on Bcl-xL overexpression. In summary, although Bcl-xL is perfectly functional in its capacity to interfere with Bax/Bak-mediated apoptosis, it did not block the Bax/Bak-mediated redistribution of nuclear proteins.

Bcl-xL overexpression does not inhibit stress-induced NPM, H1 and nucleolin redistribution. Bcl-xL cells (a) and empty vector stable transfectants (b) untreated (Con) or treated for 24 h with 25 μM cisplatin (Cis) were double stained with anti-NPM or anti-H1 together with anti-Bcl-xL antibodies, or with anti-nucleolin together with anti-FLAG antibodies, and with Hoechst 33258 (nuclei), after which they were visualized by fluorescence microscopy. The images of each treatment (left, middle and right panels) represent the same field visualized separately for detecting antibody staining (left and middle panels) and Hoechst-stained nuclei (right panel). The results presented are from a representative experiment (one of at least three independent experiments). Arrows indicate cells and their nuclei that exhibit nuclear protein redistribution. Bars, 20 μm. H1, histone 1; NPM, nucleophosmin

Bcl-xL overexpression inhibits cisplatin-induced apoptosis but not nuclear protein redistribution. Empty vector stable transfectants (pcDNA3) and Bcl-xL (Bcl-xL) MEFs untreated (Con) or treated for 24 h with 25 μM cisplatin (Cis) were double stained with anti-NPM, anti-H1, anti-Bax NT, or with anti-cytochrome c (Cyt c) together with anti-Bcl-xL antibodies, or with anti-nucleolin together with anti-FLAG antibodies. In addition, nuclei were stained with Hoechst 33258. The stained cells were visualized as described in Figure 7. The number of cells exhibiting redistribution of each of the nuclear proteins (NPM, H1 or nucleolin) (a) or apoptotic events (Bax NT exposure, cytochrome c release and apoptotic (Ap) nuclei) (b) was determined in pcDNA3 and Bcl-xL cells. The results are represented as the percentage of cells exhibiting nuclear protein redistribution or an apoptotic event from pcDNA3 or Bcl-xL (Bcl-xL-expressing (XL+) or nonexpressing (XL−)) cells (at least 100 cells). The values are represented as means±S.E.M. (bars; n=3). **P<0.01, *P<0.03, which is significantly higher than that of the corresponding untreated cells (Con) (one-tailed Student's t-test). cis, cisplatin; Con, untreated; MEFs, mouse embryonic fibroblasts; NPM, nucleophosmin; NT, N-terminal; WT, wild type
Re-expression of Bax or Bak in Bax/Bak DKO MEFs restores the nuclear protein redistribution effect
It is possible that we did not observe stress-induced nuclear protein redistribution in Bax/Bak DKO MEFs because these cells lost their responsiveness toward this process during their clonal selection in vivo or ex vivo. To clarify this point, we transiently re-introduced Bax or Bak in the form of GFP- or HA-tagged fusion proteins into Bax/Bak DKO MEFs and assessed the redistribution of NPM, H1 and nucleolin 24 h later. As a control, cells were transfected with the GFP vector. It should be noted that transfecting cells with Bax or Bak produced an apoptotic stimulus per se, so that no additional drug was needed to effectively induce apoptosis. As shown in Figure 9a, most of the Bax/Bak DKO cells that re-express GFP–Bax exhibited redistribution of NPM, H1 and nucleolin. This redistribution was not because of cell destruction, as it occurred even in cells appearing healthy (Figure 9a). Quantification of the percentage of NPM, H1 and nucleolin redistribution in GFP- or GFP–Bax-transfected cells (Figure 9b) revealed that, whereas GFP alone induced a moderate redistribution of NPM, H1 and nucleolin (34, 22 and 22%, respectively, compared with untreated Bax/Bak DKO cells (19, 9 and 3%, respectively (Figure 5d))), this effect was drastically increased by GFP–Bax re-expression (64, 70 and 83%, respectively). These results indicate that the redistribution effect was a direct consequence of the action of Bax. In addition, as mentioned above for cisplatin-treated WT MEFs (Supplementary Figure S4), the general caspase inhibitor, Boc, was unable to prevent the redistribution of nuclear proteins when it was added to GFP–Bax-transfected cells (Figure 9b). We also examined the ability of Bak to restore the redistribution effect in DKO MEFs and found that re-expressing HA–Bak induced a similar nucleolin redistribution as induced by HA–Bax (Figure 9c). Collectively, these results confirm that the redistribution of nuclear proteins is not an arbitrary side effect of stressed cells; it is mechanistically and causally linked to Bax and Bak expression.

Re-expression of Bax or Bak MEFs restores nuclear protein redistribution in Bax/Bak DKO MEFs. Bax/Bak DKO MEFs were transiently transfected with GFP, GFP–Bax, HA–Bax or HA–Bak expression vector in the presence or absence of Boc or Q-VD-OPH. After 24 h, the cells were stained and visualized as described in Figure 1. The images shown (a) are Boc-treated (100 μM) GFP–Bax transfectants. Each row represents the same field visualized separately for detecting GFP–Bax-expressing cells (left panel), nuclear protein expression (middle panel) and nuclei (right panel). The results presented are from a representative experiment (one of at least three independent experiments). Arrows indicate cells expressing GFP–Bax, which exhibit nuclear protein redistribution and their nuclei. Note the appearance of NPM in the cytosol, and the reduced intensity of H1 and nucleolin staining in the marked cells. (b) Quantification of the number of cells exhibiting nuclear protein redistribution in GFP or GFP–Bax-expressing cells. Bax/Bak DKO MEFs were transfected with GFP or GFP–Bax in the presence or absence of Boc and were analyzed as described in (a). The results shown are expressed as the percentage of cells exhibiting both nuclear protein redistribution and GFP or GFP–Bax expression, from the total population of GFP or GFP–Bax-expressing cells (at least 100 cells). Values are represented as means±S.E.M. (bars; n=3). *P<0.01, which is significantly higher than that of the corresponding controls, for example, cells transfected with GFP or with GFP and treated with Boc (one-tailed Student's t-test). (c) Quantification of the number of cells exhibiting nucleolin redistribution in GFP, HA–Bax or HA–Bak-expressing cells. Bax/Bak DKO MEFs were transfected with GFP, HA–Bax and HA–Bak expression vectors in the presence of Q-VD-OPH (20 μM). After 24 h, the cells were double stained with anti-nucleolin and anti-HA antibodies (in the HA–Bax and HA–Bak transfectants) or only with anti-nucleolin antibodies (in the GFP transfectants) together with Hoechst 33258, and visualized by fluorescence microscopy. The results shown are expressed as the percentage of cells exhibiting nucleolin redistribution and GFP, HA–Bax or HA–Bak expression, from the total population of GFP, HA–Bax or HA–Bak-expressing cells (at least 100 cells). The values shown represent the means±S.E.M. (bars; n=3). *P<0.0001, which is significantly higher than that of the GFP-expressing cells (one-tailed Student's t-test). Bars, 20 μm. DKO, double knock out; GFP, green fluorescent protein; HA, hemagglutinin; MEFs, mouse embryonic fibroblasts; NPM, nucleophosmin
The BH3 mimetic, ABT-737, induces NPM redistribution in WT, but not in DKO MEFs
BH3-only proteins are crucial signaling intermediates between apoptotic stimuli and Bax/Bak activation. To assess whether BH3-only proteins were implicated in the redistribution effect as well, we tested the action of ABT-737 (a BH3 mimetic known to effectively activate Bax and Bak25, 26) on NPM redistribution in WT and Bax/Bak DKO MEFs. Similar to previously reported studies,27 ABT-737 triggered a low (29% at 10 μM) but significant (P=0.03; Student's t-test) amount of cell death in WT, but not in Bax/Bak DKO MEFs (Supplementary Figure S7a). Furthermore, ABT-737 induced a 2.5-fold increase in NPM redistribution in WT, but not in Bax/Bak DKO MEFs (Supplementary Figure S7b). These results show that the redistribution of nuclear proteins is linked to an ABT-737-mediated Bax/Bak activation step that might involve BH3-only proteins.
Discussion
This study identifies a new function of Bax and Bak, namely, the regulation of stress-induced nuclear protein redistribution, a process proposed to have an important role in apoptosis.28 This function of Bax and Bak differs from their canonical activity on MOM perforation and cytochrome c release because it is not blocked by Bcl-xL overexpression and seems to be uncoupled from conformational changes in the N-termini of Bax and Bak.
We focused on the redistribution of three nuclear proteins, namely, NPM, H1 and nucleolin in response to four different apoptotic stimuli (cisplatin, camptothecin, doxorubicin and staurosporine). In all cases, we detected a redistribution of these proteins. This effect was observed early after inducing apoptosis. For example, substantial nuclear protein redistribution was evident at 9 h after cisplatin or camptothecin treatment, when phosphatidylserine translocation, Bax/Bak NT exposure, cytochrome c or caspase-3 activation had not yet been detected. These findings explain why the redistribution effect was independent of the Apaf-1/caspase-9 apoptosome and of caspases in general, as these components are known to be activated later. Collectively, our results suggest that the redistribution effect occurs upstream or independently of the mitochondrial pathway.
Not all nuclear proteins exhibit nuclear protein redistribution. For example, KAP-1 did not change its nuclear localization under the same conditions. This indicates that the redistribution effect was specific for a certain class of nuclear proteins that share a yet unknown property. As the redistribution precedes the appearance of apoptotic features and did not affect all nuclear proteins, it cannot be because of a general leakage from destroyed nuclei.
It was previously proposed that cytosolic H1.2 and NPM facilitate apoptosis through Bax/Bak.13, 14, 15, 16 Our finding that the H1 and NPM redistribution is mediated through Bax/Bak implies that Bax and Bak act upstream of H1.2 and NPM, and thus regulate the ability of these nuclear proteins to activate them. The role of nucleolin redistribution in response to stress signals is more enigmatic. Nucleolin is a shuttling protein. It may regulate apoptosis by modulating the import and/or export of nucleolar components. In addition, nucleolin can reach the cell membrane and thus affect cell adhesion and proliferation.18, 19, 29 Importantly, nucleolin stabilizes Bcl-2 and Bcl-xL mRNAs,22, 30, 31 and suppresses p53 translation and induction after DNA damage.32 Redistribution of nucleolin may, therefore, promote a prosurvival effect.
The conclusion that the stress-induced redistribution of NPM, H1 and nucleolin is controlled by Bax and Bak is based on the results showing that (i) MEFs deficient in these two proteins did not exhibit the redistribution effect; (ii) the BH3 mimetic ABT-737, which is known to act through Bax/Bak, also induced the redistribution effect in a Bax/Bak-dependent manner; and (iii) re-expression of Bax or Bak in Bax/Bak DKO cells restored the redistribution effect. It is, however, important to note that nuclear protein redistribution can also occur independently of Bax/Bak, as untreated DKO MEFs exhibited a low, but detectable level of spontaneous redistribution, and stresses induced by transfection or by staurosporine treatment (the latter only in the case of H1) triggered a moderate redistribution effect.
The question arises as to how would Bax/Bak regulate nuclear/cytoplasmic redistribution/transport from their site of action (mitochondria, ER and nucleus). We suspect that they may trigger a yet unidentified caspase- and apoptosome-independent signaling pathway, which either increases the permeability of the nuclear pore, thereby enabling diffusion of specific nuclear proteins or impairs active nuclear transport by affecting the distribution of mobile transport factors. The latter assumption is supported by a previous study showing that proapoptotic insults induced the redistribution of nuclear transport factors such as the small GTPase, Ran, which determines the direction of transport across the nuclear envelope.33
Two findings of our study were unexpected. First, nuclear redistribution was mediated by Bax/Bak, but occurred before cells showed Bax/Bak NT exposure. The two processes may even inhibit each other, because the probability that nuclear protein redistribution was observed together with Bax/Bak NT exposure in the same cell was lower than would be anticipated if these two events were random. Second, nuclear redistribution was not inhibited by Bcl-xL overexpression. In this regard, it is worth noting, however, that ABT-737 did trigger (although at a low amount) nuclear protein redistribution. We currently do not know how this BH3 mimetic, which is thought to act by binding to Bcl-2 family proteins such as Bcl-xL,25, 26 induces nuclear protein redistribution in a Bax/Bak-dependent and apparently Bcl-xL-nonblockable manner. One possibility is that at the high concentration of ABT-737 that is required to kill normal cells (e.g., MEFs), ABT-737 also may act through a Bax/Bak-dependent mechanism that is not inhibited by Bcl-xL. Alternatively, ABT-737 might directly activate Bax/Bak, and thereby not only provoke apoptosis through Bax/Bak NT exposure but also induce nuclear protein redistribution through another Bax/Bak-dependent mechanism. Nonetheless, our findings suggest that the redistribution effect is not mediated by the canonical Bax/Bak pore-forming activity on the MOM. The pore-forming activity of Bax/Bak may still mediate the redistribution effect, but on another subcellular compartment such as the nucleus. Consistent with this idea, it was shown that anti- and proapoptotic Bcl-2 family proteins can reside in the nucleus, on the nuclear membrane or at the nuclear pore.34, 35 Alternatively, Bax/Bak can mediate the redistribution effect from the endoplasmic reticulum (by regulating intracellular calcium levels)36 or from the cytosol/mitochondria by affecting mitochondrial fusion and fission.37 Further studies are required to distinguish between these mechanisms.
Despite intensive research and vast progress during the last decade, the mechanism whereby Bax and Bak promote their proapoptotic effects is far from being solved. Although there is compelling evidence that the main action of Bax and Bak is to render MOM perforation inhibitable by Bcl-2/Bcl-xL, other modus operandi may exist for the two proteins. Our results suggest that Bax and Bak may also contribute to apoptosis by regulating nuclear protein redistribution and that this effect might be mediated by a new, yet unknown, apoptotic signaling pathway.
Materials and Methods
Materials
All reagents used were purchased from Sigma (St. Louis, MO, USA) unless otherwise stated. Boc and Z-VAD-FMK were purchased from ICN Biomedicals (Irvine, CA, USA). Q-VD-OPH was purchased from BioVision Research Products (Mountain View, CA, USA). ABT-737 was synthesized as described by Oltersdorf et al.25
Cell culture
Primary WT, Bax−/−, Bak−/−, Bax/Bak DKO, caspase-9−/− and Apaf-1−/− MEFs were obtained from Andreas Strasser (The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia). They were immortalized by the 3T9 method38 and grown in high-glucose Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal calf serum. WT and WT1 immortalized MEFs were generated from two independent primary cultures of WT MEFs, each obtained from different embryos. Different MEFs were treated with or without the indicated apoptotic triggers. When the effect of caspase inhibitors was tested, the inhibitors were applied 1 h before the addition of cisplatin.
Plasmids
The expression vectors used in this study were pEGFP (Clontech Laboratories, Mountain View, CA, USA); pEGFP–Bax (GFP–Bax);39 pcDNA3 HA–Bax (HA–Bax);40 FLAG–Bcl-xL;41 pcDNA3 HA–Bak (HA–Bak) (a gift from Gabriel Nuñez, Michigan Medical School, Ann Arbor, MI, USA); pEGFP–nucleolin (GFP–nucleolin)42 (a gift from Ronit Pinkas-Kramarski, Tel Aviv University); and pEGFP–B23 (GFP–NPM)43 (provided by Dennis Discher, University of Pennsylvania).
Transfection
Transfection into Bax/Bak DKO cells was carried out using jetPEI transfection reagent (Polyplus Transfection SA, Illkirch, France) or with lipofectamine (Invitrogen Life Technologies, Paisley, UK), according to the manufacturer's instructions. jetPEI was used in the experiments depicted in Figure 9a and lipofectamine was used in all other transfections. One day before transfection, the cells were seeded at a density of 105 cells per well in 12-well plates. When transfections were performed in the presence of Boc, it was added 5 h after adding the reagents for transfection. The ratios for the different DNA vectors were 1 : 1 pEGFP, GFP–Bax, HA–Bax or HA–Bak pcDNA3. To generate Bcl-xL, pcDNA3 and GFP–nucleolin cell lines, WT MEFs were transfected with FLAG–Bcl-xL, pcDNA3 or pEGFP–nucleolin expression vector, and stable transfectants were selected using 1 mg/ml geneticin (G418).
Immunofluorescence staining
The different MEFs were grown in 12-well plates, 105 cells per dish, on 18-mm cover slips coated with collagen. After treatments, cells were fixed and stained with different antibodies and Hoechst 33258 dye, as described previously.44 Next, the cells were incubated with the following primary antibodies: mouse anti-nucleophosmin (Zymed Laboratories, San Francisco, CA, USA), mouse anti-histone H1(AE-4), rabbit anti-nucleolin C23 (H-250) (both from Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti-KAP-1 (BD Transduction Laboratories, Lexington, KY, USA), rabbit anti-Bak-NT, rabbit anti-Bax-NT (both from Upstate Biotechnology, Lake Placid, NY, USA), anti-cytochrome c (BD Biosciences, San Jose, CA, USA) or anti-active caspase-3 (CM1 rabbit polyclonal antibodies (Idun Pharmaceuticals, La Jolla, CA, USA)). Fluorescent images were captured using a fluorescence microscope (Nikon ECLIPSE TE2000-S, Nikon Instruments Inc., Melville, NY, USA) connected to a CCD camera (CoolSNAP HQ, RoPER Scientific, Trenton, NJ, USA) with plan Apo VC × 60/1.4 oil emersion objective using Image-Pro PLUS (version 4.5.1; Media Cybernetics Inc, Bethesda, MD, USA) software and exported to Photoshop (Adobe). To determine the number of cells exhibiting the redistribution effect or apoptotic features, the fluorescent stained cells were counted under the fluorescence microscope.
Cell viability assay: His–GFP–annexin-V/PI FACS analysis
The different MEFs were seeded on 60-mm dishes (106 cells per dish). After 24 h, the cells were treated with the apoptotic drug (25 μM cisplatin, 1 μM camptothecin, 1 μM doxorubicin or 100 nM staurosporine). For assessing apoptosis, His–GFP–annexin-V/PI labeling was carried out as described45 with minor modifications. Briefly, after treatment, the cells were detached, centrifuged, washed with PBS and then incubated for 15 min at room temperature in a 50-μl annexin-V binding buffer containing 3 μg/ml His–GFP–annexin-V, followed by the addition of 400 μl annexin-V binding buffer and 50 μg/ml propidium iodide. Finally, the cells were subjected to FACS analysis using Becton Dickinson FaCSort (BD Biosciences). The data were analyzed using the WinMDI program supplied by the manufacturer.
Assay for DEVDase activity
The activity of caspases was measured in terms of the assayed DEVDase activity as described previously,46 with minor modifications. WT MEFs (2 × 104 cells) were seeded in 96-well plates and treated with cisplatin (25 μM) with or without 100 μM Boc. After 24 h, the medium was removed and the cells were washed with PBS and then lysed using 50 μl of lysis buffer per well containing 50 mM Tris-HCl (pH 8.0), 120 mM NaCl, 5 mM EDTA and 0.5% Nonidet-P40, for 10 min at 37 °C. Thereafter, 50 μl of Ac-DEVD-7AMC (Biomol Research Laboratories, Plymouth Meeting, PA, USA) solution containing 50 μM Ac-DEVD-7AMC, 40 mM HEPES (pH 7.5), 20% glycerol and 4 mM DTT was added to each well and fluorescence was measured after incubation (at 37 °C) for an additional 30 min at an excitation wavelength of 340 nm and emission wavelengths of 460 nm.
Statistical analysis
Data were expressed as mean values±S.E.M. Statistical evaluation was determined by one-tailed Student's t-test. A value of P<0.05 was considered statistically significant. To examine whether the redistribution effect (e.g., of H1) and NT exposure (e.g., Bax) are independent events, we determined independently the number of cells exhibiting H1 redistribution; the number of cells exhibiting Bax NT exposure; the number of cells exhibiting Bax NT from those cells showing H1 redistribution; and the number of cells exhibiting H1 redistribution from those cells showing Bax NT. Under the null hypothesis of independence, we expect that the proportion of cells exhibiting, for example, Bax NT exposure in the total cell population (P(BaxNT)) will equal their proportion in the sub-population of cells exhibiting H1 redistribution (P(BaxNT/H1)), and the proportion of cells exhibiting H1 exposure in the total cell population (P(H1)) will equal their proportion in the sub-population of cells exhibiting BaxNT exposure (P(H1)/BaxNT). We then calculated the χ2 value while comparing the proportion of cells exhibiting Bax NT in the total cell population with their proportion in the sub-population of cells showing H1 redistribution. We also calculated the χ2 value while comparing the proportion of cells exhibiting H1 redistribution in the total cell population with their proportion in the sub-population of cells showing Bax NT exposure. The two χ2 values were summed and their significance was calculated under two degrees of freedom.
Abbreviations
- Boc:
-
BOC-Asp(OMe)-FMK
- Bax/Bak DKO:
-
Bax−/− Bak−/− double knockout
- H1:
-
histone H1
- MEFs:
-
mouse embryonic fibroblasts
- MOM:
-
mitochondrial outer membrane
- NT:
-
N terminal
- NPM:
-
nucleophosmin
- Q-VD-OPH:
-
quinoline-Val-Asp(OME)-CH2-PH
- WT:
-
wild type
- Z-VAD-FMK:
-
N-benzyloxycarbonyl-Val-Ala-Asp(O-methyl)-fluoromethylketone
References
Youle RJ, Strasser A . The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9: 47–59.
Borner C . The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Mol Immunol 2003; 39: 615–647.
Lindsten T, Thompson CB . Cell death in the absence of Bax and Bak. Cell Death Differ 2006; 13: 1272–1276.
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292: 727–730.
Scorrano L, Korsmeyer SJ . Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochem Biophys Res Commun 2003; 304: 437–444.
Sharpe JC, Arnoult D, Youle RJ . Control of mitochondrial permeability by Bcl-2 family members. Biochim Biophys Acta 2004; 1644: 107–113.
Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 2003; 11: 577–590.
Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004; 303: 1010–1014.
Lin B, Kolluri SK, Lin F, Liu W, Han YH, Cao X et al. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell 2004; 116: 527–540.
Paroni G, Henderson C, Schneider C, Brancolini C . Caspase-2 can trigger cytochrome c release and apoptosis from the nucleus. J Biol Chem 2002; 277: 15147–15161.
Grisendi S, Mecucci C, Falini B, Pandolfi PP . Nucleophosmin and cancer. Nat Rev Cancer 2006; 6: 493–505.
Yung BY, Chang FJ, Luo KJ . Dipyridamole enhancement of doxorubicin-induced translocation of nucleophosmin and inhibition of cell growth in HL-60 cells. Int J Cancer 1991; 49: 592–597.
Konishi A, Shimizu S, Hirota J, Takao T, Fan Y, Matsuoka Y et al. Involvement of histone H1.2 in apoptosis induced by DNA double-strand breaks. Cell 2003; 114: 673–688.
Okamura H, Yoshida K, Amorim BR, Haneji T . Histone H1.2 is translocated to mitochondria and associates with Bak in bleomycin-induced apoptotic cells. J Cell Biochem 2008; 103: 1488–1496.
Kerr LE, Birse-Archbold JL, Short DM, McGregor AL, Heron I, Macdonald DC et al. Nucleophosmin is a novel Bax chaperone that regulates apoptotic cell death. Oncogene 2007; 26: 2554–2562.
Thompson J, Finlayson K, Salvo-Chirnside E, MacDonald D, McCulloch J, Kerr L et al. Characterisation of the Bax–nucleophosmin interaction: the importance of the Bax C-terminus. Apoptosis 2008; 13: 394–403.
Vignali M, Workman JL . Location and function of linker histones. Nat Struct Biol 1998; 5: 1025–1028.
Ginisty H, Sicard H, Roger B, Bouvet P . Structure and functions of nucleolin. J Cell Sci 1999; 112 ( Pt 6): 761–772.
Mongelard F, Bouvet P . Nucleolin: a multiFACeTed protein. Trends Cell Biol 2007; 17: 80–86.
Calle A, Ugrinova I, Epstein AL, Bouvet P, Diaz JJ, Greco A . Nucleolin is required for an efficient herpes simplex virus type 1 infection. J Virol 2008; 82: 4762–4773.
Kito S, Morimoto Y, Tanaka T, Haneji T, Ohba T . Cleavage of nucleolin and AgNOR proteins during apoptosis induced by anticancer drugs in human salivary gland cells. J Oral Pathol Med 2005; 34: 478–485.
Otake Y, Sengupta TK, Bandyopadhyay S, Spicer EK, Fernandes DJ . Retinoid-induced apoptosis in HL-60 cells is associated with nucleolin down-regulation and destabilization of Bcl-2 mRNA. Mol Pharmacol 2005; 67: 319–326.
Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R et al. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 1998; 94: 739–750.
Ruffolo SC, Shore GC . BCL-2 selectively interacts with the BID-induced open conformer of BAK, inhibiting BAK auto-oligomerization. J Biol Chem 2003; 278: 25039–25045.
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435: 677–681.
Chen S, Dai Y, Harada H, Dent P, Grant S . Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res 2007; 67: 782–791.
van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006; 10: 389–399.
Ferrando-May E . Nucleocytoplasmic transport in apoptosis. Cell Death Differ 2005; 12: 1263–1276.
Srivastava M, Pollard HB . Molecular dissection of nucleolin's role in growth and cell proliferation: new insights. FASEB J 1999; 13: 1911–1922.
Soundararajan S, Chen W, Spicer EK, Courtenay-Luck N, Fernandes DJ . The nucleolin targeting aptamer AS1411 destabilizes Bcl-2 messenger RNA in human breast cancer cells. Cancer Res 2008; 68: 2358–2365.
Zhang J, Tsaprailis G, Bowden GT . Nucleolin stabilizes Bcl-X L messenger RNA in response to UVA irradiation. Cancer Res 2008; 68: 1046–1054.
Takagi M, Absalon MJ, McLure KG, Kastan MB . Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005; 123: 49–63.
Ferrando-May E, Cordes V, Biller-Ckovric I, Mirkovic J, Gorlich D, Nicotera P . Caspases mediate nucleoporin cleavage, but not early redistribution of nuclear transport factors and modulation of nuclear permeability in apoptosis. Cell Death Differ 2001; 8: 495–505.
Mandal M, Adam L, Mendelsohn J, Kumar R . Nuclear targeting of Bax during apoptosis in human colorectal cancer cells. Oncogene 1998; 17: 999–1007.
Quignon F, De Bels F, Koken M, Feunteun J, Ameisen JC, de The H . PML induces a novel caspase-independent death process. Nat Genet 1998; 20: 259–265.
Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 2003; 300: 135–139.
Suen DF, Norris KL, Youle RJ . Mitochondrial dynamics and apoptosis. Genes Dev 2008; 22: 1577–1590.
Todaro GJ, Green H . Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol 1963; 17: 299–313.
Schinzel A, Kaufmann T, Schuler M, Martinalbo J, Grubb D, Borner C . Conformational control of Bax localization and apoptotic activity by Pro168. J Cell Biol 2004; 164: 1021–1032.
Lindenboim L, Yuan J, Stein R . Bcl-xS and Bax induce different apoptotic pathways in PC12 cells. Oncogene 2000; 19: 1783–1793.
Kaufmann T, Schlipf S, Sanz J, Neubert K, Stein R, Borner C . Characterization of the signal that directs Bcl-x(L), but not Bcl-2, to the mitochondrial outer membrane. J Cell Biol 2003; 160: 53–64.
Di Segni A, Farin K, Pinkas-Kramarski R . Identification of nucleolin as new ErbB receptors-interacting protein. PLoS One 2008; 3: e2310.
Dundr M, Misteli T, Olson MO . The dynamics of postmitotic reassembly of the nucleolus. J Cell Biol 2000; 150: 433–446.
Lindenboim L, Kringel S, Braun T, Borner C, Stein R . Bak but not Bax is essential for Bcl-xS-induced apoptosis. Cell Death Differ 2005; 12: 713–723.
Egger L, Schneider J, Rheme C, Tapernoux M, Hacki J, Borner C . Serine proteases mediate apoptosis-like cell death and phagocytosis under caspase-inhibiting conditions. Cell Death Differ 2003; 10: 1188–1203.
Naumann U, Wischhusen J, Weit S, Rieger J, Wolburg H, Massing U et al. Alkylphosphocholine-induced glioma cell death is BCL-X(L)-sensitive, caspase-independent and characterized by massive cytoplasmic vacuole formation. Cell Death Differ 2004; 11: 1326–1341.
Acknowledgements
We thank Professor Andreas Strasser for providing the various apoptotic protein-deficient MEFs, Professor Pinkas-Kramarski for providing the GFP–nucleolin plasmid, as well as Ms. Ayelet Levy and Ms. Ilana Gelernter for statistical analysis. This work was supported by the German–Israeli Foundation (to RS and CB); the Cancer Biology Research Center (CBRC) of Tel Aviv University (to RS); the Deutsche Forschungsgemeinschaft (DFG) (BO-1933) and the Spemann Graduate School of Biology and Medicine (SGBM) (GSC-4) funded by the DFG (to CB); as well as the German Carreras Leukemia Foundation (to CB, DJCLS R 06/09).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by L Scorrano
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Supplementary information
Rights and permissions
About this article
Cite this article
Lindenboim, L., Blacher, E., Borner, C. et al. Regulation of stress-induced nuclear protein redistribution: a new function of Bax and Bak uncoupled from Bcl-xL. Cell Death Differ 17, 346–359 (2010). https://doi.org/10.1038/cdd.2009.145
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.145
Keywords
This article is cited by
-
LINC complex protein nesprin-2 has pro-apoptotic activity via Bcl-2 family proteins
Cell Death Discovery (2024)
-
Apoptotic stress induces Bax-dependent, caspase-independent redistribution of LINC complex nesprins
Cell Death Discovery (2020)
-
The nuclear envelope: target and mediator of the apoptotic process
Cell Death Discovery (2020)
-
Non-canonical function of Bax in stress-induced nuclear protein redistribution
Cellular and Molecular Life Sciences (2013)
-
The secrets of the Bcl-2 family
Cell Death & Differentiation (2012)
