
Abstract
RBPjk-dependent Notch signaling regulates both the onset of chondrocyte hypertrophy and the progression to terminal chondrocyte maturation during endochondral ossification. It has been suggested that Notch signaling can regulate Sox9 transcription, although how this occurs at the molecular level in chondrocytes and whether this transcriptional regulation mediates Notch control of chondrocyte hypertrophy and cartilage development is unknown or controversial. Here we have provided conclusive genetic evidence linking RBPjk-dependent Notch signaling to the regulation of Sox9 expression and chondrocyte hypertrophy by examining tissue-specific Rbpjk mutant (Prx1Cre;Rbpjkf/f ), Rbpjk mutant/Sox9 haploinsufficient (Prx1Cre;Rbpjkf/f;Sox9f/+ ), and control embryos for alterations in SOX9 expression and chondrocyte hypertrophy during cartilage development. These studies demonstrate that Notch signaling regulates the onset of chondrocyte maturation in a SOX9-dependent manner, while Notch-mediated regulation of terminal chondrocyte maturation likely functions independently of SOX9. Furthermore, our in vitro molecular analyses of the Sox9 promoter and Notch-mediated regulation of Sox9 gene expression in chondrogenic cells identified the ability of Notch to induce Sox9 expression directly in the acute setting, but suppresses Sox9 transcription with prolonged Notch signaling that requires protein synthesis of secondary effectors.
Similar content being viewed by others


Introduction
The limb skeleton is derived via the process of endochondral ossification, which begins with the condensation of mesenchymal progenitor cells within the developing limb-buds. Cells within condensations undergo chondrogenesis, creating a cartilage template of the skeletal elements, while cells at the periphery known as perichondrial cells ultimately differentiate into osteoblasts that form bone. Chondrocytes of the developing skeletal elements proliferate with round disorganized chondroctyes near the epiphyses giving rise to flattened chondrocytes organized in columns near the metaphyses. This organization provides directionality to the longitudinal expansion of cartilage rudiments. As chondrocytes approach the center of the elements, they exit the cell cycle and begin the process of hypertrophic differentiation. Chondrocyte hypertrophy is a key step during endochodral bone development where chondrocytes dramatically alter their morphology and size to generate a mineralizing cartilage matrix. Hypertrophic chondrocytes also secrete molecules important in inducing osteoblastogenesis, recruiting vascular tissue to the primary ossification center, and aiding in the process of replacing the mineralized cartilage with bone.1 When this process is perturbed, chondrodysplasias and known cartilage and skeletal disorders arise.
Specific transcription factors are responsible for directing the differentiation pattern of chondrocytes and osteoblasts during endochondral ossification. SOX9 is a transcription factor known to be a master regulator of chondrogenesis and the differentiated chondrocyte phenotype.2 In humans, heterozygous mutations of Sox9 results in campomelic dysplasia, a lethal developmental disorder characterized by generalized hypoplasia of endochondral bones and XY sex reversal.3,4 In mice and humans, haploinsufficiency of Sox9 is lethal shortly after birth and phenocopies many of the skeletal anomalies present in campomelic dysplasia.5 A study of Sox9−/− chimeric mice, as well as a study using Prx1Cre to selectively inactivate Sox9 floxed alleles within the skeletogenic mesenchyme has identified Sox9 as necessary for the formation of mesenchymal condensations.6,7 SOX9 activation of chondrocyte-specific genes is partially mediated through its activation of two other distantly related members of the SOX family, SOX5 and SOX6.8 The three proteins, together, have been shown to activate numerous cartilage-specific extracellular matrix genes, including Col2a1, Col11a2, Matrilin-1, Col27a1, and Agc12 that maintain the differentiated chondrocyte environment. Recent studies have also shown that over-expression of Sox9 in hypertrophic chondrocytes results in delayed terminal chondrocyte maturation and cartilage matrix turnover, suggesting a specific role in regulating chondrocyte hypertrophy.9 While previous studies have shown negative regulation of Sox9 by the β-catenin signaling pathway results in accelerated chondrocyte hypertrophy10 and positive regulation by the TGFb/BMP signaling pathways induces chondrogenesis,11,12 the precise genetic mechanisms regulating Sox9 gene expression during cartilage development remain ill-defined.
Recent studies have identified the Notch signaling pathway as a potential regulator of Sox9 in chondrocytes. Briefly, Notch signaling is initiated when a ligand of the Delta/Jagged families bind to a Notch receptor (NOTCH1-4). Binding initiates a series of cleavage events, culminating in gamma-secretase mediated release of the intracellular portion of the Notch receptor, known as the Notch Intracellular Domain (or NICD). Once released, NICD translocates to the nucleus where it binds the transcriptional repressor RBPjk, converting it to a transcriptional activator. A transcriptionally active complex composed, in part, of NICD, RBPjk and MAML drives expression of target genes, such as those of the Hes/Hey gene family.13,14 In the absence of NICD, RBPjk recruits the co-repressors SMRT and SHARP resulting in a suppression of transcription.15,16 Recently, two studies have shown that over-expression of NICD, using either the Prx1Cre to target the skeletogenic mesenchyme or the Col2Cre to target chondro-osteoprogenitor cells, results in inhibition of condensation formation, significantly reduced levels of Sox9, and, ultimately, an inability to form skeletal elements.17,18 These mutants are markedly similar to those observed in mutant embryos lacking Sox9 expression in the skeletogenic mesenchyme.6 Furthermore, mutant mice with over-expression of Sox9 in hypertrophic chondrocytes using the Col10a1 promoter9 are histologically very similar to previously reported mice lacking various components of the Notch signaling pathway, including the Notch1 and Notch2 receptors and Rbpjk.19,20 Interestingly, loss of Rbpjk in chondro-osteoprogenitor cells leads to increased levels of Sox9 gene expression in growth plate chondrocytes, specifically in the hypertrophic cells, while over-expression of NICD leads to decreased Sox9 expression throughout the growth plate.18 These data suggest a possible mechanism, by which RBPjk-dependent Notch signaling is required to modulate Sox9 expression during cartilage development and chondrocyte hypertrophy.
To examine the genetic interaction between RBPjk-dependent Notch signaling and Sox9 we analyzed tissue-specific Rbpjk mutant (Prx1Cre;Rbpjkf/f ), Rbpjk mutant/Sox9 haploinsufficient (Prx1Cre;Rbpjkf/f;Sox9f/+ ), and control embryos for alterations in chondrocyte hypertrophy and cartilage growth. Additionally, chondrogenic cell lines were used to assess the specific mechanisms by which Notch signaling regulates the Sox9 promoter. The cartilage and molecular phenotypes observed in our genetic studies combined with our in vitro molecular data demonstrate that RBPjk-dependent Notch signaling coordinates the onset of chondrocyte maturation and cartilage growth largely via indirect regulation of Sox9.
Materials and methods
Mouse strains
All mouse strains including Prx1Cre, Rbpjkf/f , and Sox9f/f are as previously described.5,21,22 Prx1Cre and Sox9f/f mice were obtained from the Jackson Laboratory; Rbpjκf/f mice were a generous gift from Dr. Tasuku Honjo (Kyoto Graduate School of Medicine, Japan). Embryos of the genotypes Prx1Cre;Rbpjkf/+ , Prx1Cre;Rbpjkf/+;Sox9f/+ , Prx1Cre;Rbpjkf/f , Prx1Cre;Rbpjkf/f;Sox9f/+ , and Cre-negative littermate controls were produced in Mendelian ratios to be analyzed from E14.5 to E18.5. All animal studies were performed in accordance with the guidelines set forth by the Institutional Animal Care and Use Committee.
Analyses of mouse embryos
Embryonic tissues were harvested at E14.5–E18.5, fixed in 10% neutral buffered formalin, decalcified in 14% EDTA, processed, and embedded in paraffin prior to sectioning at 6 μm. Alcian Blue/Hematoxylin/Orange G (ABH/OG) staining was performed according to standard methodologies. In situ hybridization was performed as described previously,17,19,20, 23,24 using 35S-labeled riboprobes. Immunohistochemistry for SOX9 was performed using the Vectastain Elite Rabbit IgG Kit (Vector) and Santa Cruz Biotechnology SOX9 antibody (sc20095). SOX9 antibody was prepared in 4% normal goat serum using a 1:200 dilution without antigen retrieval. Color reaction was performed using Vector ImmPACT DAB (Vector); sections were counterstained with hematoxylin (Zymed).
Whole-mount skeletal staining of embryos was performed as previously described.17,25,26
Sox9 luciferase assays
Forty-eight hours before transfection, ATDC5 cells (RIKEN) were plated in 24-well plates. ATDC5 cells were maintained at 37 °C with 5% CO2 in DMEM/F12 (1:1) supplemented with 5% fetal bovine serum, and 1% penicillin/streptomycin. Transfections were completed using FuGENE HD (Promega) with 600 ng of Flag or NICD1, 200 ng Sox9-pGL3-promoter DNA constructs, and 8 ng Renilla (Promega). Flag and NICD1 constructs were transfected first for 24 hours before adding the respective Sox9-pGL3 constructs and Renilla for 24 additional hours. After the 48 hours of transfections, cellular extracts were collected using the lysis buffer in the dual-luciferase assay (Promega). Firefly luciferase activity was assessed using 20 μL or microliter of cellular extracts followed by immediate analysis of Renilla luciferase activity. Analysis of the data was performed by normalizing Sox9-pGL3 luciferase activity to Renilla luciferase activity; normalized luciferase activity was then normalized to the Flag control of the respective Sox9-pGL3 constructs. Luciferase assays were performed in triplicate and repeated three times. Statistical analysis was performed using a two-tailed, unpaired t-test.
In vitro Notch activation and protein synthesis inhibition Six-well tissue culture plates were coated with RetroNectin (Takara) solution (20 μg·mL−1 in 1X PBS, 1 mL per well) at 4 °C overnight while gently rocking. The following day, each well was blocked with 1 mL 2% BSA (Sigma) for 30 minutes at room temperature and briefly washed with 1X PBS. The plate was then coated with anti-IgG (Sigma) (10 μg·mL−1 in 1X PBS, 1 mL per well) at 4 °C overnight while gently rocking. The following day, each well was blocked with 1 mL 2% BSA for 30 minutes at room temperature and briefly washed with 1X PBS. The plate was next coated with either IgG (Sigma) (10 μg·mL−1 in 1X PBS, 1 mL per well) or Recombinant JAG1 protein (R&D Systems) (10 μg·mL−1 in 1X PBS, 1 mL per well) at 4 °C overnight while gently rocking. The following day, each well was blocked with 1 mL 2% BSA for 30 minutes at room temperature and briefly washed with 1X PBS. Coated plates were air dried for 1 hour in the tissue culture hood, sealed with parafilm and stored at 4 °C until use. ATDC5 cells were seeded onto the IgG- or JAG1-coated plates at a density of 500 000 cells per well in ITS media ± the protein synthesis inhibitor, cycloheximide (10 μg·mL−1) (Sigma). RNA was harvested at 0, 2, 4, and 8 hours after seeding and quantitative reverse transcription polymerase chain reaction (RT-PCR) was performed to measure levels of Hes1, Hey1, and Sox9 gene expression.
Results
Loss of RBPjk leads to inappropriate expression of SOX9 in hypertrophic chondrocytes
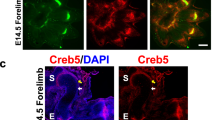
In previous studies, we have demonstrated that RBPjk-dependent Notch signaling is necessary for normal onset of chondrocyte maturation as well as terminal hypertrophy.20 In these studies, the Prx1Cre transgene was used to selectively target removal of Rbpjk from the skeletogenic mesenchyme of the developing limb. While a clear role for RBPjk-dependent Notch signaling during chondrocyte maturation was established, the downstream mechanisms have not been determined. Previous studies have shown that Sox9 is expressed in the epiphyseal resting and proliferating chondrocytes but not in the hypertrophic chondrocytes.27 Interestingly, immunohistochemistry for SOX9 reveals persistent, inappropriate expression of SOX9 protein within deeper zones of the hypertrophic region in Prx1Cre;Rbpjkf/f mutants, compared to littermate controls (Figure 1a and b). This is consistent with data showing that over-expression of NICD in chondro-osteoprogenitor and mesenchymal progenitor cells leads to reduced Sox9 gene expression in cartilage and limb-bud mesenchyme, respectively.18 Conversely, when RBPjk is removed, Sox9 gene expression is elevated in all chondrocytes.18 Collectively, these data suggest a possible mechanism for RBPjk-dependent Notch signaling regulation of chondrocyte hypertrophy via Sox9 regulation.

Loss of RBPjk leads to inappropriate expression of SOX9 in hypertrophic chondrocytes. Immunohistochemistry for SOX9 in control (a) and Prx1Cre;Rbpjkf/f (b) E14.5 humerus sections.
Sox9 haploinsufficiency rescues delays in onset of chondrocyte hypertrophy due to loss of Rbpjk
To begin to understand the genetic interactions between RBPjk-dependent Notch signaling and Sox9, we removed a single copy of Sox9 in the background of Rbpjk mutants. For completeness, the breeding strategy allowed for the generation of the following genotypes: Cre-negative controls, Rbpjk heterozygous (Prx1Cre;Rbpjkf/+ ), double heterozygous (Prx1Cre;Rbpjkf/+;Sox9f/+ ), Rbpjk mutants (Prx1Cre;Rbpjkf/f ), and Rbpjk mutant/Sox9 haploinsufficient (Prx1Cre;Rbpjkf/f;Sox9f/+ ) embryos.
Histological examination of E14.5 embryos with ABH/OG staining (Figure 2) revealed the expected delay in onset of hypertrophy in Rbpjk mutant embryos, as indicated by the shorter hypertrophic zone (Figure 2a2) compared to littermate controls (Figure 2a1). Interestingly, Rbpjk mutant/Sox9 haploinsufficient embryos (Figure 2a4) revealed a longer hypertrophic zone compared to controls, indicating acceleration in chondrocyte hypertrophy. The longer hypertrophic zone was also seen in double heterozygous embryos (Figure 2a3), suggesting that Sox9 haploinsufficiency is sufficient to accelerate chondrocyte hypertrophic differentiation. To ensure that the accelerated hypertrophic phenotype was due to Sox9 haploinsufficiency and not a result of double heterozygosity of Rbpjk and Sox9, we examined Sox9 heterozygous embryos (Prx1Cre;Sox9f/+ ). At E14.5, loss of a single allele of Sox9 results in a longer hypertrophic zone compared to littermate controls (Figure 2a5 and a6). Finally, quantitative analysis of the hypertrophic zone revealed a significantly shorter hypertrophic zone in Rbpjk mutants compared to controls, while the Rbpjk mutant/Sox9 haploinsufficient hypertrophic zones were significantly longer than both controls and Rbpjk mutants and were similar to Rbpjk/Sox9 double heterozygous mice (Figure 2b1). Removal of a single Rbpjk allele in the Sox9 haploinsufficient background had no significant effect on the Sox9 heterozygous change in hypertrophy (Figure 2b1 and b2). Similar histological changes in the onset of chondrocyte hypertrophy were observed in all elements examined from both forelimb and hindlimb (data not shown).

Notch regulates the onset of chondrocyte hypertrophy via Sox9. (a) Histological analyses of control (a1), Prx1Cre;Rbpjkf/f (a2), Prx1Cre;Rbpjkf/+;Sox9f/+ (a3), Prx1Cre;Rbpjkf/f;Sox9f/+ (a4), as well as, control (a5) and Prx1Cre;Sox9f/+ embryonic tibia sections at E14.5. (b) Quantification of the lengths of the hypertrophic zone, expressed as a percentage of the total length of the element. (b1) WT – wild-type, Rbpjk (Prx1Cre;Rbpjkf/f ), Dbl Het (Prx1Cre;Rbpjkf/+;Sox9f/+ ), RBPJk; Sox9 (Prx1Cre;Rbpjkf/f;Sox9f/+ ). (b2) WT – wild-type, Sox9 Het (Prx1Cre;Sox9f/+ ).
Cartilage elements of the limb skeleton were further analyzed for common markers of chondrocyte hypertrophy using in situ hybridization (Figure 3). As observed in Figure 2, histological analysis by ABH/OG staining revealed a smaller hypertrophic zone in Rbpjk mutant embryos, but a larger hypertrophic zone in Rbpjk mutant/Sox9 haploinsufficient embryos (Figure 3a–c). As we previously reported,20 molecular analysis using in situ hybridization revealed significantly smaller Ihh and Col10a1 domains in Rbpjk mutant embryos compared to controls (Figure 3d, e, g, h). Furthermore, molecular analysis for Mmp13 showed a small number of cells expressing Mmp13 in the control sections (Figure 3j, orange circle), while no Mmp13 expressing cells are detected in Rbpjk mutant sections (Figure 3k). Conversely, in situ hybridization for Ihh and Col10a1 reveals larger expression domains in Rbpjk mutant/Sox9 haploinsufficient embryos, and a wide domain of Mmp13 expressing cells (Figure 3f, i, l). These data indicate that during chondrocyte hypertrophy, Sox9 is likely downstream of RBPjk-dependent Notch signaling such that a reduction in Sox9 expression in Rbpjk mutants largely corrects or over-corrects the delay in chondrocyte hypertrophy.

Notch regulates the onset of chondrocyte hypertrophy via Sox9. Histological and molecular analysis of control, Prx1Cre;Rbpjkf/f and Prx1Cre;Rbpjkf/f;Sox9f/+ embryonic tibia sections at E14.5. ABH/OG staining (a, b, c). In situ hybridization for markers of chondrocyte maturation – Indian Hedgehog (Ihh) (d, e, f), Collagen 10a1 (Col10a1) (g, h, i) and Matrix Metaloproteinase 13 (Mmp13) (j, k, l). Yellow arrowheads indicate primary Ihh expressing domains. Yellow double-headed arrows indicate Col10a1 expression domain. Orange circle highlights Mmp13 expression.
Cartilage element shortening due to Sox9 haploinsufficiency is rescued by loss of Rbpjk
Examination of ABH/OG stained full-length tibia sections (Figure 4a) revealed that skeletal elements of the Rbpjk mutants (Figure 4a2), the double heterozygous (Figure 4a3) and the Rbpjk mutant/Sox9 haploinsufficient embryos (Figure 4a4) were all significantly smaller than littermate controls and displayed bowed skeletal elements (Figure 4a1). Double heterozygous mutants displayed the shortest skeletal elements, which were comparable to Sox9 heterozygous mutant embryos (Prx1Cre;Sox9f/+ ) in both length and curvature of the elements (Figure 4a3 and 4b2). Quantification revealed that the tibias of all three mutants were significantly shorter than controls, and that the double heterozygous were significantly shorter than Rbpjk mutants or the Rbpjk mutant/Sox9 haploinsufficient embryos (Figure 4c). Interestingly, removal of both Rbpjk floxed alleles in the Sox9 haploinsufficient background restores tibia length equivalent to the Rbpjk mutant size, although does not correct the curvature or bowing of the Sox9 heterozygous or double heterozygous mutants. Furthermore, BrdU analyses showed no significant difference in chondrocyte proliferation between all mutant genotypes (data not shown), suggesting the rescued length in Rbpjk mutant/Sox9 haploinsufficient embryos as compared to double heterozygous or Sox9 heterozygous mutants is likely due to alterations in hypertrophic differentiation. Collectively, these data suggest that complete removal of Rbpjk-dependent Notch signaling elevates Sox9 expression to a level that can counteract some of the chondrogenic effects oberserved in Sox9 happloinsuficient mutant mice.

Cartilage element length reduction due to Sox9 haploinsufficiency is rescued by loss of RBPjk-dependent Notch signaling. (a) ABH/OG staining of control (a1), Prx1Cre;Rbpjkf/f (a2), Prx1Cre;Rbpjkf/+ ;Sox9f/+ (a3) and Prx1Cre;Rbpjkf/f ;Sox9f/+ (a4) E18.5 full length tibia sections. (b) ABH/OG staining of control (b1) and Prx1Cre;Sox9f/+ (b2) E18.5 tibia sections. (c) Quantification of the size difference of E18.5 tibia sections.
Sox9 haploinsufficiency does not rescue delays in chondrocyte terminal hypertrophy due to loss of Rbpjk
We further analyzed limbs from E18.5 embryos in a similar manner to those from E14.5. ABH/OG staining of proximal tibia growth plates revealed the long hypertrophic zone characteristic of the delayed terminal chondrocyte hypertrophy observed in Rbpjk mutants, compared to controls (Figure 5a1 and a2). Interestingly, the double heterozygous embryos appeared to have relatively normal growth plates without any significant change in length of the hypertrophic zone, however, the overall size of the cartilage growth plate was slightly smaller than controls (Figure 5a3). Interestingly, the growth plate and hypertrophic zone of the Rbpjk mutant/Sox9 haploinsufficient embryos phenocopied that of Rbpjk mutants, specifically in regard to the expanded hypertrophic zone (Figure 5a2 and a4). In situ hybridization for chondrocyte hypertrophy markers, Ihh, Col10a1, and Mmp13, was performed on E18.5 tibia sections. Analysis of the proximal growth plate revealed expanded domains of Ihh, Col10a1, and Mmp13 in both the Rbpjk mutants and Rbpjk mutant/Sox9 haploinsufficient embryos compared to controls, while the double heterozygous embryos reveal domains similar in size to controls with a mild enhancement in Mmp13 (Figure 5a4–a12). Consistent with the results observed in Figure 1, IHC results demonstrate that SOX9 protein persists deeper into the hypertrophic zone of Rbpjk mutants due to the enhanced Sox9 expression as compared to controls (Figure 5b1 and b2). Interestingly, while SOX9 persistence within the hypertrophic chondrocytes was reduced in Rbpjk mutant/Sox9 haploinsufficient embryos compared to Rbpjk mutants, the progression through terminal chondrocyte hypertrophy was still delayed (identified by the expanded hypertrophic zones) (Figure 5b2 and b3). These data indicate that RBPjk-dependent Notch signaling regulation of terminal chondrocyte hypertrophy and cartilage matrix turnover may function independent of Sox9 transcriptional control.

Notch regulation of terminal chondrocyte maturation is not likely to be mediated via Sox9. (a) Histological and molecular analysis of control, Prx1Cre;RBPjkf/f, Prx1Cre;RBPjkf/f; Sox9f/+ embryonic tibia sections at E18.5. ABH/OG staining (a1–a3). Markers of chondrocyte maturation – Indian Hedgehog (Ihh) (a4–a6), Collagen 10a1 (Col10a1) (a7–a9) and Matrix Metaloproteinase 13 (Mmp13) (a10–a12) were analyzed by in situ hybridization. (b) Immunohistochemistry for SOX9 in control (b1), Prx1Cre;RBPjkf/f (b2), and Prx1Cre;RBPjkf/f ;Sox9f/+ (b3) embryonic tibia sections at E18.5. Yellow double-headed arrows indicate the length of the SOX9 expressing hypertrophic chondrocyte domain. Red double-headed arrows indicate the total length of the hypertrophic zone.
Notch mediated suppression of Sox9 requires secondary effectors
The Notch signaling pathway has been shown to be an important regulator of Sox9, although the mechanism of this regulation is controversial. Here, we performed a rigorous analysis of the known Sox9 promoter and Sox9 gene expression in the context of Notch signaling activation. To achieve this, we first utilized Sox9 luciferase constructs containing various sizes of the Sox9 promoter. As shown in Figure 6a, the only construct containing RBPjk consensus sequences is the 6.8 kb known promoter fragment. Using ATDC5 cells, we co-transfected the 6.8 kb (6.8 kb WT) construct with either Flag (control) or NICD1 over-expression plasmids. As expected, the over-expression of NICD1 suppressed Sox9 luciferase activity (Figure 6b). According to Chen et al., the RBPjk consensus sequence, located 3 kb upstream of the transcriptional start site, is the site significantly enriched of the NICD/RBPjk complex.28 To determine if this site is responsible for the suppression of Sox9, we made point mutations designed to prevent the binding of RBPjk to its consensus sequence (TGGGAA to TCCGAA).29 After co-transfecting the 6.8 kb mutant (6.8 kb MT) construct with Flag or NICD1, we observed suppression of Sox9 luciferase activity despite the RBPjk mutation (Figure 6b). To rule out the possibility that NICD may be binding to another RBPjk site in the 6.8 kb promoter region, we utilized a Sox9 luciferase construct containing only 1 kb of promoter sequence upstream of the transcriptional start site, which does not contain any RBPjk binding sites. We co-transfected the 1 kb Sox9 construct with Flag or NICD1, and observed a similar level of suppression as seen with the 6.8 kb construct (Figure 6c). Interestingly, most or all of the Notch-mediated suppression of Sox9 luciferase activity was lost when the promoter fragments were reduced to contain only 0.5 kb to 0.32 kb of the Sox9 promoter (Figure 6c). These data provide evidence that the NICD1 suppression of Sox9 likely occurs via secondary effectors and does not utilize the RBPjk binding sites in a suppressive manner.

Notch signaling inhibits Sox9 gene expression via secondary effectors. (a) Diagram for the localization of a RBPjk consensus binding site (yellow box is wild-type sequence and blue box is mutant sequence) and N-box consensus binding site (red box) in the Sox9 promoter and luciferase constructs. Core consensus sequences are listed in diagram. (b) ATDC5 cells were co-transfected with Flag or NICD1 over-expression vectors and either a wild-type 6.8 kb Sox9-Luciferase complex (−6.8 kb WT) or 6.8 kb construct with a mutated RBPjk binding site (−6.8 kb MT). Luciferase levels were measured 24 hours after transfection. (c) ATDC5 cells were co-transfected with Flag or NICD1 over-expression vectors and either a 1 kb, 0.5 kb, or 0.32 kb Sox9-Luciferase deletion constructs. Luciferase levels were measured 24 hours after transfection. (d) Quantitative RT-PCR assessing Hes1 (d1, d2), Hey1 (d3, d4), and Sox9 (d5, d6) gene expression in ATDC5 cells at 0-, 2-, 4-, and 8-hour post-culture on IgG versus JAG1 coated plates in the absence (d1, d3, d5) or presence (d2, d4, d6) of cycloheximide.
To elucidate the mechanism of the Notch suppression of Sox9, we used a bioinformatic approach to analyze the 1 kb upstream fragment (TRANSFAC; Biobase). Interestingly, we located a conserved N-box consensus sequence (CACCAG) (Figure 6a) at −681 to −676. The N-box is one of two known binding sites of the HES/HEY family of transcription factors. HES/HEY factors are well characterized as downstream Notch target genes. Upon binding to the N-box, HES/HEY factors will recruit co-repressors inhibiting gene transcription.30 Additionally, it has been shown in the developing limb that Notch signaling can induce Hes/Hey gene expression.17 Interestingly, the 0.5 kb and 0.32 kb Sox9 luciferase constructs utilized in Figure 6c both lack the identified N-box. These constructs are deficient in suppressing Sox9 luciferase expression suggesting that Notch suppression of Sox9 is mediated via secondary effectors that likely include one or more of the HES/HEY factors.
To further determine whether Notch inhibits Sox9 expression directly or via downstream target gene activation, we initiated Notch signaling in ATDC5 cells using Jagged1 (JAG1) coated plates and assessed Notch-induced gene expression (Figure 6d). Cultures were maintained in the presence or absence of the protein synthesis inhibitor, cycloheximide, in order to assess whether translation of downstream Notch target genes is required for Sox9 gene regulation. RNA was isolated from cultures at 0, 2, 4, and 8 hours of culture, and quantitative RT-PCR was performed to measure levels of the established Notch target genes, Hes1 and Hey1,31 as well as Sox9. The culture of ATDC5 cells on JAG1 coated plates versus IgG coated control plates leads to significant up-regulation of both Hes1 and Hey1 gene expression across all time points (Figure 6d1–d4). When we conducted this experiment in the presence of cycloheximide (Figure 6d2 and d4), similar levels of Hes1 and Hey1 up-regulation were observed indicating that both Hes1 and Hey1 transcription are directly activated by RBPjk-dependent Notch signaling, as has been previously reported.31 Interestingly, when we examine Sox9 transcriptional regulation in the absence of cycloheximide, we see an initial mild up-regulation of Sox9 gene expression at 2 hours, but then see a sustained inhibition of Sox9 expression at 4 and 8 hours (Figure 6d5). When we assess Notch signaling effects on Sox9 gene expression in the presence of cycloheximide, we find the same but significant mild up-regulation of Sox9 at 2 hours. However, the inhibition of Sox9 gene expression seen in the absence of cycloheximide at 4 and 8 hours (Figure 6d5) is lost when ATDC5 cells are cultured in the presence of cycloheximide and JAG1 activation (Figure 6d6). These results indicate that Notch signaling can initially promote Sox9 gene expression directly through the NICD/RBPjk transcriptional activating complex, but the inhibition of Sox9 requires the translation of Notch-dependent target genes.
Discussion
Our previous work identified RBPjk-dependent Notch signaling as a necessary regulator of both the onset of chondrocyte hypertrophy and the progression to terminal chondrocyte maturation, although the mechanism remains unknown.20 Here we have provided conclusive genetic evidence linking RBPjk-dependent Notch signaling to the regulation of Sox9 expression and chondrocyte hypertrophy. In Rbpjk mutant embryos, haploinsufficiency of Sox9 was able to rescue the delays in onset of chondrocyte hypertrophy, but had little if any impact on delayed progression to terminal chondrocyte maturation and cartilage matrix turnover. Interestingly, limb length reductions characteristic of Sox9 haploinsufficiency were partially resolved when RBPjk-dependent Notch signaling was completely removed, suggesting that a delicate regulation and balance of Sox9 expression is required to coordinate chondrocyte hypertrophy and cartilage growth. Furthermore, our in vitro results demonstrate the ability of Notch signaling to acutely enhance Sox9 expression likely through direct RBPjk-dependent regulation, while continuous Notch signaling suppresses Sox9 via secondary effectors.
SOX9 is well established as the master regulator of chondrogenesis and a factor required for the maintenance of the immature chondrocyte phenotype,5–7,32,33 and thus a potential target of RBPjk-dependent Notch function in cartilage development. Our work, as well as others, has shown that Sox9 is normally down-regulated in hypertrophic chondrocytes,27 indicating that Sox9 down-regulation is required for the onset of hypertrophy. A recent study has specifically addressed this by using a BAC-Col10a1 promoter to drive continuous expression of Sox9 in hypertrophic chondrocytes.9 Maintenance of SOX9 within hypertrophic chondrocytes results in an elongated zone of hypertrophic chondrocytes at E18.5, strikingly similar to the elongated hypertrophic zone caused by the delayed progression to terminal chondrocyte maturation observed in Notch mutant embryos.19,20 While Hattori et al.9 did not examine the onset of chondrocyte hypertrophy, at E14.5, using their Sox9 over-expressing mutants; our Prx1Cre transgene induced Sox9 heterozygous mutant mice demonstrate accelerated hypertrophy via the down-regulation of Sox9. Therefore, our data are consistent with a specific role for SOX9 in regulating the onset of chondrocyte hypertrophy. Based on these data and our understanding of Notch signaling, we explored the genetic relationship between Notch signaling and Sox9 in the context of chondrocyte hypertrophy. While the delayed onset of chondrocyte hypertorphy due to loss of RBPjk could be fully rescued by the removal of a single allele of Sox9, the same was not true concerning delayed terminal chondrocyte maturation. As we have suggested, it is therefore not likely that Sox9 is involved in RBPjk-dependent Notch regulation of terminal chondrocyte hypertrophy, however, we cannot rule out the possibility that removal of a single allele of Sox9 was not significant enough of a knock-down to effect the terminal chondrocyte maturation phenotype. In our current genetic model utilizing the Prx1Cre, removal of both Sox9 alleles was impossible, as SOX9 is required for condensation formation and early chondrogenesis.6
Our data demonstrate that haploinsufficiency of Sox9 results in significant shortening and bowing of the endochondral elements. These data are consistent with previously reported Sox9 mutants, including Prx1Cre;Sox9fx/+ embryos analyzed by Akiyama et al., which displayed hypoplasia and bowing of various endochondral elements.6 Global haploinsufficiency of Sox9 results in more severe limb element hypoplasia and bowing as compared to the Prx1Cre targeted Sox9 mutants.5 Interestingly, at E18.5, Sox9 global heterozygous embryos display an approximately two-fold increase in the length of their hypertrophic zone. This expansion is likely due to early onset of hypertrophy, as opposed to the delays in terminal chondrocyte hypertrophy observed in Notch loss-of-function mutants.5,18–20 An extension of the hypertrophic zone due to early onset in chondrocyte hypertrophy has been previously reported in β-catenin GOF embryos,34 which has also been shown to down-regulate SOX9 during the transition to chondrocyte hypertrophy by specifically targeting SOX9 protein for proteosomal degradation.35 Here, we demonstrated that complete removal of RBPjk-dependent Notch signaling in the Sox9 haploinsufficient background was enough to restore some Sox9 expression and promote cartilage growth similar to that observed in Rbpjk mutants, but not enough to correct the bowing observed in all Sox9 haploinsufficient mutants.
In recent work using the Col2Cre transgene to study RBPjk-dependent and –independent Notch signaling effects on vertebral formation, Chen et al. proposed a model whereby RBPjk-dependent Notch signaling suppression of Sox9 is important for normal axial skeletogenesis.36 Controversially, they proposed a mechanism by which the NICD-RBPjk complex directly inhibits Sox9 gene expression via direct binding to the Sox9 promoter. While provocative, there is no evidence to suggest NICD-RBPjk complexes can function as transcriptional repressors since NICD recruitment to RBPjk and DNA is known to displace transcriptional repressors ultimately leading to transcriptional activation. Chen et al. provided chromatin immunoprecipitation (ChIP) assays detecting the recruitment of the NICD-RBPjk complex to RBPjk binding sites in the Sox9 promoter as evidence of their claim. While it is possible and probable that NICD-RBPjk complexes bind to the Sox9 promoter, it is more likely that NICD-RBPjk may have a physiological role in directly promoting Sox9 expression in specific contexts. This is supported by data that demonstrate the ability of Notch to induce Sox9 gene expression in embryonic stem cells, pancreatic cells, and Müller glial cells.37–39 Furthermore, the work presented here in chondrogenic cells demonstrates that site specific mutations of the RBPjk binding site (proposed by Chen et al. as the repressive element) does not yield any change in Notch-mediated suppression of Sox9. Alternatively, we propose that Notch signaling leads to an indirect suppression of Sox9 via the induction of alternative downstream targets. This is also supported by our own data, which demonstrates that Notch-induced inhibition of Sox9 in chondrogenic cells requires active protein synthesis. We therefore hypothesize that one or more of the HES/HEY family of repressive bHLH transcription factors, some of the primary downstream targets of RBPjk-dependent Notch signaling, act as mediators of Notch signaling to repress Sox9 gene expression. This is further supported by the loss of Notch-mediated suppression of Sox9 driven luciferase activity when the Sox9 promoter was truncated to exclude a putative HES/HEY binding site. Further studies will be required to elucidate the exact role HES/HEY factors may play in specifically regulating Sox9 expression and cartilage development.
Authors’ contribution
Authors’ roles: Study design: Matthew J. Hilton. Study conduct: Anat Kohn, Timothy P. Rutkowski, Zhaoyang Liu, and Anthony J. Mirando. Data collection: Anat Kohn, Timothy P. Rutkowski, and Zhaoyang Liu. Data analysis: Anat Kohn, Timothy P. Rutkowski, and Zhaoyang Liu. Data interpretation: Anat Kohn, Timothy P. Rutkowski, Zhaoyang Liu, Regis J. O’Keefe, Matthew J. Hilton, and Michael J. Zuscik. Drafting manuscript: Anat Kohn, Timothy P. Rutkowski, Zhaoyang Liu, Regis J. O’Keefe, Matthew J. Hilton, and Michael J. Zuscik. Approving final version of manuscript: Anat Kohn, Timothy P. Rutkowski, Zhaoyang Liu, Regis J. O’Keefe, Matthew J. Hilton, and Michael J. Zuscik. Anat Kohn, Timothy P. Rutkowski, Zhaoyang Liu, and Matthew J. Hilton take responsibility for the integrity of the data analysis.
References
Kronenberg HM . Developmental regulation of the growth plate. Nature 2003; 423: 332–336.
Akiyama H, Lefebvre V . Unraveling the transcriptional regulatory machinery in chondrogenesis. J Bone Miner Metab 2011; 29: 390–395.
Foster JW, Dominguez-Steglich MA, Guioli S et al. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature 1994; 372: 525–530.
Wagner T, Wirth J, Meyer J et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell 1994; 79: 1111–1120.
Bi W, Huang W, Whitworth DJ et al. Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc Natl Acad Sci U S A 2001; 98: 6698–6703.
Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B . The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev 2002; 16: 2813–2828.
Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B . Sox9 is required for cartilage formation. Nat Genet 1999; 22: 85–89.
Lefebvre V, Li P, de Crombrugghe B . A new long form of Sox5 (L-Sox5), Sox6 and Sox9 are coexpressed in chondrogenesis and cooperatively activate the type II collagen gene. EMBO J 1998; 17: 5718–5733.
Hattori T, Muller C, Gebhard S et al. SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development 2010; 137: 901–911.
Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C . Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell 2005; 8: 727–738.
Wan M, Cao X . BMP signaling in skeletal development. Biochem Biophys Res Commun 2005; 328: 651–657.
Yoon BS, Lyons KM . Multiple functions of BMPs in chondrogenesis. J Cell Biochem 2004; 93: 93–103.
Engin F, Lee B . NOTCHing the bone: insights into multi-functionality. Bone 2010; 46: 274–280.
Kopan R, Ilagan MX . The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 2009; 137: 216–233.
Bray SJ . Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 2006; 7: 678–689.
Chiba S . Notch signaling in stem cell systems. Stem Cells 2006; 24: 2437–2447.
Dong Y, Jesse AM, Kohn A et al. RBPjkappa-dependent Notch signaling regulates mesenchymal progenitor cell proliferation and differentiation during skeletal development. Development 2010; 137: 1461–1471.
Mead TJ, Yutzey KE . Notch pathway regulation of chondrocyte differentiation and proliferation during appendicular and axial skeleton development. Proc Natl Acad Sci U S A 2009; 106: 14420–14425.
Hilton MJ, Tu X, Wu X et al. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat Med 2008; 14: 306–314.
Kohn A, Dong Y, Mirando AJ et al. Cartilage-specific RBPjkappa-dependent and -independent Notch signals regulate cartilage and bone development. Development 2012; 139: 1198–1212.
Han H, Tanigaki K, Yamamoto N et al. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int Immunol 2002; 14: 637–645.
Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ . Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 2002; 33: 77–80.
Hilton MJ, Tu X, Cook J, Hu H, Long F . Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development 2005; 132: 4339–4351.
Hilton MJ, Tu X, Long F . Tamoxifen-inducible gene deletion reveals a distinct cell type associated with trabecular bone, and direct regulation of PTHrP expression and chondrocyte morphology by Ihh in growth region cartilage. Dev Biol 2007; 308: 93–105.
Hu H, Hilton MJ, Tu X, Yu K, Ornitz DM, Long F . Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development 2005; 132: 49–60.
McLeod MJ . Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology 1980; 22: 299–301.
Zhao Q, Eberspaecher H, Lefebvre V, De Crombrugghe B . Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev Dyn 1997; 209: 377–386.
Chen S, Tao J, Bae Y et al. Notch gain of function inhibits chondrocyte differentiation via Rbpj-dependent suppression of Sox9. J Bone Miner Res 2013; 28: 649–659.
Tun T, Hamaguchi Y, Matsunami N, Furukawa T, Honjo T, Kawaichi M . Recognition sequence of a highly conserved DNA binding protein RBP-J kappa. Nucleic Acids Res 1994; 22: 965–971.
Fischer A, Gessler M . Delta-Notch--and then? Protein interactions and proposed modes of repression by Hes and Hey bHLH factors. Nucleic Acids Res 2007; 35: 4583–4596.
Bray S, Bernard F . Notch targets and their regulation. Curr Top Dev Biol 2010; 92: 253–275.
Akiyama H, Kim JE, Nakashima K et al. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc Natl Acad Sci U S A 2005; 102: 14665–14670.
Lefebvre V, de Crombrugghe B . Toward understanding SOX9 function in chondrocyte differentiation. Matrix Biol 1998; 16: 529–540.
Dao DY, Jonason JH, Zhang Y et al. Cartilage-specific beta-catenin signaling regulates chondrocyte maturation, generation of ossification centers, and perichondrial bone formation during skeletal development. J Bone Miner Res 2012; 27: 1680–1694.
Akiyama H, Lyons JP, Mori-Akiyama Y et al. Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev 2004; 18: 1072–1087.
Chen S, Tao J, Bae Y et al. Notch gain of function inhibits chondrocyte differentiation via Rbpj-dependent suppression of Sox9. J Bone Miner Res 2013; 28: 649–659.
Haller R, Schwanbeck R, Martini S et al. Notch1 signaling regulates chondrogenic lineage determination through Sox9 activation. Cell Death Differ 2012; 19: 461–469.
Muto A, Iida A, Satoh S, Watanabe S . The group E Sox genes Sox8 and Sox9 are regulated by Notch signaling and are required for Muller glial cell development in mouse retina. Exp Eye Res 2009; 89: 549–558.
Seymour PA, Shih HP, Patel NA et al. A Sox9/Fgf feed-forward loop maintains pancreatic organ identity. Development 2012; 139: 3363–3372.
Acknowledgements
This work was supported in part by the following United States National Institute of Health grants: R01 grants (AR057022 and AR063071), R21 grant (AR059733 to MJH), a P30 Core Center grant (AR061307), and a T32 training grant that supported both AK and TPR (AR053459 to Regis J. O’Keefe and Michael J. Zuscik). The NICD and FLAG control plasmids were a gift from Dr. Raphael Kopan (Cincinnati Children’s Hospital) and the 6.8 kb Sox9-Luciferase plasmid was a kind gift from Dr. Peter Koopman (University of Queensland). We would like to gratefully acknowledge the technical expertise and assistance of Sarah Mack, Kathy Maltby, and Ashish Thomas within the Histology, Biochemistry, and Molecular Imaging Core in the Center for Musculoskeletal Research at the University of Rochester Medical Center.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution NonCommercial-NoDerivs 4.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Kohn, A., Rutkowski, T., Liu, Z. et al. Notch signaling controls chondrocyte hypertrophy via indirect regulation of Sox9. Bone Res 3, 15021 (2015). https://doi.org/10.1038/boneres.2015.21
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/boneres.2015.21
This article is cited by
-
MYL3 protects chondrocytes from senescence by inhibiting clathrin-mediated endocytosis and activating of Notch signaling
Nature Communications (2023)
-
Lymphoid enhancer-binding factor-1 promotes stemness and poor differentiation of hepatocellular carcinoma by directly activating the NOTCH pathway
Oncogene (2019)
-
Differential tissue specific, temporal and spatial expression patterns of the Aggrecan gene is modulated by independent enhancer elements
Scientific Reports (2018)
-
Molecular characterization of mesenchymal stem cells in human osteoarthritis cartilage reveals contribution to the OA phenotype
Scientific Reports (2018)
-
Transcription factor profiling identifies Sox9 as regulator of proliferation and differentiation in corneal epithelial stem/progenitor cells
Scientific Reports (2018)
