
Abstract
The hypoxia inducible factors (Hifs) are evolutionarily conserved transcriptional factors that control homeostatic responses to low oxygen. In developing bone, Hif-1 generated signals induce angiogenesis necessary for osteoblast specification, but in mature bone, loss of Hif-1 in osteoblasts resulted in a more rapid accumulation of bone. These findings suggested that Hif-1 exerts distinct developmental functions and acts as a negative regulator of bone formation. To investigate the function of Hif-1α in osteoanabolic signaling, we assessed the effect of Hif-1α loss-of-function on bone formation in response to intermittent parathyroid hormone (PTH). Mice lacking Hif-1α in osteoblasts and osteocytes form more bone in response to PTH, likely through a larger increase in osteoblast activity and increased sensitivity to the hormone. Consistent with this effect, exposure of primary mouse osteoblasts to PTH resulted in the rapid induction of Hif-1α protein levels via a post-transcriptional mechanism. The enhanced anabolic response appears to result from the removal of Hif-1α-mediated suppression of β-catenin transcriptional activity. Together, these data indicate that Hif-1α functions in the mature skeleton to restrict osteoanabolic signaling. The availability of pharmacological agents that reduce Hif-1α function suggests the value in further exploration of this pathway to optimize the therapeutic benefits of PTH.
Similar content being viewed by others


Introduction
Parathyroid hormone (PTH), an 84-amino-acid polypeptide, is an essential regulator of mineral homeostasis and bone remodeling. Released by the chief cells of the parathyroid gland in response to deviations in serum calcium levels, PTH primarily acts on kidney and bone to increase calcium reabsorption and liberate calcium from bone matrix, respectively.1 Additionally, intermittent PTH administration is recognized for its anabolic effects in bone, and the first 34 amino acids of the hormone form the basis for the only Food and Drug Administration-approved anabolic agent to treat osteoporosis.2–4
The anabolic actions of PTH have been extensively studied in laboratory rodent models.5 Through its actions on the PTH receptor,6 expressed by osteoblasts and osteocytes, histological analyses suggest that intermittent PTH increases bone acquisition by increasing the number of bone-forming osteoblasts.7–9 More recent molecular analyses have attempted to identify signaling mechanisms and components that allow PTH to reduce bone cell apoptosis,9 stimulate progenitor cell recruitment10 and activate formerly quiescent bone lining cells.11 In addition to the activation of cyclic AMP (cAMP) and protein kinase A signaling,12 components of the insulin-like growth factor pathway,13–15 the transforming growth factor-β pathway10,16 and Wnt/β-catenin signaling17,18 have all been demonstrated to be required for the full osteo-anabolic response to PTH. The identification of factors or signaling mechanisms that inhibit bone formation after PTH administration has been less common,19,20 but examining such mechanisms could facilitate the development of strategies to increase the therapeutic efficacy of intermittent PTH.
Hypoxia inducible factor-1 (Hif-1) is most widely recognized for its role in the cellular response to molecular oxygen levels.21,22 A basic helix–loop–helix transcription factor, the activity and cellular abundance of Hif-1 is regulated by an oxygen-dependent proteolysis mechanism. At normal oxygen tensions, the α-subunit of the protein (Hif-1α) undergoes prolyl hydroxylation, which initiates recognition by the von Hippel–Lindau (Vhl) tumor suppressor protein, a component of the E3 ubiquitin ligase that targets Hif-1α for proteasomal degradation. When oxygen tensions fall below 5%, prolyl hydroxylation is inhibited; Hif-1α accumulates and translocates to the nucleus where it forms a dimer with the Hif-1β subunit. In vitro studies suggest that Hif-1 regulates the expression of several hundred genes involved in angiogenic and metabolic responses,21 and utilizes both direct promoter binding23 as well as indirect mechanisms to alter gene expression.24
Within bone, Hif-1 exerts distinct developmental functions. In developing bone, Hif-1 generated signals are required for angiogenesis, which appears to be necessary for initial specification of bone-forming osteoblasts. Consistent with this idea, mice lacking Hif-1α in osteoblasts and osteocytes develop poorly vascularized bones with reduced cortical and trabecular bone volume, while Hif overexpression results in highly vascularized and dense bone.25,26 As Hif-1α mutant mice mature, a second inhibitory function emerges such that Hif-1 acts as a negative regulator of bone formation. In this regard, cortical and trabecular bone volume normalize with age and Hif-1α mutants are more sensitive to mechanical stimuli.27
In this study, we investigated the function of Hif-1α in osteo-anabolic signaling by assessing the effect of Hif-1α loss-of-function on bone formation in response to parathyroid hormone. In addition to hypoxia, Hif-1α expression is induced by a number of stimuli and signaling pathways critical for normal osteoblast function, including some that are used by PTH to increase bone formation.27–29 Moreover, PTH stimulates vascular remodeling in bone,30 which suggests two potential mechanisms by which Hif-1 might impact PTH-induced anabolism. Here, we demonstrate that PTH administration results in Hif-1α expression by osteoblasts both in vivo and in vitro, and that by interacting with β-catenin, Hif-1α suppresses the anabolic response. As a result, the removal of Hif-1α from osteoblasts and osteocytes sensitizes bone to PTH treatment by enhancing the activity of osteoblasts. These findings indicate that Hif-1α is a more general suppressor of osteo-anabolic signaling and acts to inhibit signals beyond those associated with mechanical loading.
Materials and methods
Generation of transgenic mice
The generation of mice lacking Hif-1α in osteoblasts and osteocytes (ΔHif-1α) was described previously.26,27 Briefly, OC-Cre mice31 were crossed with mice in which the second exon of Hif-1α is floxed.32 Mice containing Hif-2α-floxed alleles,33 Vhl-floxed alleles34 and mTOR-floxed (mTOR: mammalian target of rapamycin) alleles35 have been described previously. All mice were maintained on a C57BL/6 background. PCR analysis from ear or tail biopsies was used to confirm genotypes. The Institutional Animal Care and Use Committee of the Johns Hopkins University School of Medicine approved all animal procedures.
Administration of human PTH in vivo
Female ΔHif-1α and control mice were grown until 10 weeks of age at which point daily (7 day per week) subcutaneous injections of 100 µL vehicle or human PTH 1–34 (Bachem Inc., Torrance, CA, USA) were initiated. PTH concentrations (20 µg·kg−1 or 40 µg·kg−1) were adjusted weekly based on body mass measurements. All mice were sacrificed at 16 weeks of age. Blood samples were collected at sacrifice for analysis of serum markers of bone resorption and formation. Serum was collected and immediately stored at −80 °C. Serum concentrations of C-terminal telopeptide (RatLaps; IDS Inc., Scottsdale, AZ, USA) and N-terminal propeptide of type 1 procollagen (P1NP; IDS Inc.) were determined via commercially available ELISA. Two additional groups of female ΔHif-1α and control mice were treated with PTH (40 μg·kg−1 subcutaneous) for 4 or 16 h and then sacrificed. Femurs were dissected and prepared for immunohistochemical analysis of Hif-1α expression (sc-10790; Santa Cruz, Dallas, TX, USA) according to standard techniques or homogenized in TRIzol (Invitrogen, Grand Island, NY, USA) for RNA analysis after flushing the bone of marrow.
Skeletal analysis
To examine bone architecture, the mouse femur was scanned using a desktop microtomographic imaging system (Skyscan 1172; Skyscan, Kontich, Belgium) in accordance with the recommendations of the American Society for Bone and Mineral Research.36 The femur was scanned at 50 keV and 200 mA using a 0.5 mm aluminum filter with an isotropic voxel size of 10 µm. The resulting two-dimensional images are shown in gray scale. Trabecular bone parameters were assessed in the distal femur 500 μm proximal to the growth plate and extending for 2 mm (200 CT slices). Cortical bone parameters were assessed at the femoral midshaft and represent an average of 50 CT slices (500 µm). Dynamic measures of bone formation were assessed by injection of two sequential 0.2 mL doses of calcein (0.8 mg·mL−1) delivered 3 and 10 days prior to sacrifice. The femur was fixed in ethanol, dehydrated and embedded in methylmethacrylate. Three micron sections were cut with a Microm microtome and stained with Mason-Goldner trichrome stain. The number of osteoblasts and osteoclasts per bone perimeter were measured at standardized sites under the growth plate at a magnification of ×200 using a semi-automatic method (Osteoplan II; Kontron, Munich, Germany). These parameters comply with the guidelines of the nomenclature committee of the American Society for Bone and Mineral Research.37,38
Osteoblast isolation and culture
Osteoblasts were isolated from the calvaria of newborn Hif-1α-floxed, Hif-2α-floxed, mTOR-floxed and Vhl-floxed mice by serial digestion in 1.8 mg·mL−1 collagenase type I and maintained in α-MEM (minimum essential medium, alpha modification) supplemented with 10% FBS (Fetal bovine serum) and 1% penicillin/streptomycin. To disrupt Hif-1α, Hif-2α, mTOR or Vhl expression, osteoblasts were infected with control adenovirus expressing green fluorescent protein or adenovirus expressing Cre recombinase (Vector Biolabs, Philadelphia, PA, USA) at an MOI (multiplicity of infection) of 100. Osteoblasts were harvested 48 h after adenoviral infection and deletion efficiency was assessed in a portion of the cell population by real-time PCR. The remaining cells were replated for stimulation with PTH. Pharmacological agents were obtained from Sigma Aldrich, dissolved in DMSO and added to cell cultures with appropriate vehicle controls 30–60 min before PTH treatments.
Quantitative real-time PCR and chromatin immunoprecipitation
Total RNA was extracted from osteoblasts or homogenized femurs using TRIzol (Invitrogen) and 1 µg was reverse transcribed using the iScript cDNA synthesis system (Bio-Rad, Hercules, CA, USA). Two microliters of cDNA was subjected to PCR amplification using the iQ SYBR Green Supermix (Bio-Rad). Primer sequences were obtained from PrimerBank (http://pga.mgh.harvard.edu/primerbank/index.html). Reactions were normalized to endogenous β-actin reference transcript.
Chromatin immunoprecipitation assays were performed using an Agarose ChIP Kit (Pierce, Rockford, IL, USA) according to the manufacturer’s instructions and a ChIP-qualified antibody specific for β -catenin (PAS-16192; Thermo Scientific, Waltham, MA, USA). Precipitated DNA (2 µL) was subjected to PCR amplification by qPCR and normalized to reference reactions utilizing input DNA. Primer sequences for the Axin2 promoter are available upon request.
Protein isolation and assays
Protein was extracted from cultured osteoblasts in 0.1% Triton X-100 containing protease and phosphatase inhibitors. The extracts were separated on 10% SDS/polyacrylamide gels and transferred to PVDF (polyvinyl difluoride) membranes. Antibodies for Hif-1α (NB100–105), Hif-2α (NB100–122) and Hif-1β (NB100–124) were obtained from Novus Biological. Antibodies for phospho-Akt (S473, 9721), Akt (9272), phospho-p70 S6 kinase (9206), p70 S6 kinase (9202), phospho-Creb (9198), Creb (9197), phospho-Erk (9101), Erk (9107) and β -catenin (2698) were obtained from Cell Signaling Technologies, Danvers, MA, USA. Bound antibodies were visualized using either the Supersignal West Femto or West Pico Substrates (Pierce). Co-immunoprecipitation was performed overnight at 4 °C in a reaction containing 2 µg of antibody specific for Hif-1α (Novus) or β-catenin (Cell Signaling Technologies, San Diego, CA, USA).
Statistical analysis
Results are expressed as mean±s.e.m. All statistical tests were two-sided. A P-value less than 0.05 was considered significant. Comparability of two groups of data was assessed using a Student’s t-test.
Results
PTH stimulates Hif-1α expression in osteoblasts
Mice that lack Hif-1α in osteoblasts and osteocytes (Hif-1αflox/flox; Oc-CreTG/+, hereafter referred to as ΔHif-1α) exhibit early deficits in both cortical and trabecular bone architecture that are at least partially attributable to impairments in skeletal vascularization.25,26 As the mutant mice mature, bone architecture normalizes,27 indicating that Hif-1α exerts distinct developmental functions and likely acts to suppress osteo-anabolic signaling. To assess this inhibitory function, we examined the influence of Hif-1α on the response of osteoblasts to PTH.
To establish that Hif-1α regulates osteo-anabolic signaling in response to PTH, we first examined the ability of the hormone to induce the expression of Hif-1α in osteoblasts. In cultures of normoxic calvarial osteoblasts, PTH (10 nmol·L−1) rapidly increased Hif-1α protein levels, with expression levels peaking between 2 and 4 h after stimulation and remaining elevated through 8 h of treatment (Figure 1a). Protein levels of Hif-2α and Hif-1β were not affected. However, desferoxamine, an iron chelator that inhibits the activity of the prolyl-hydroxylase enzymes that initiate the targeting of Hif-α subunits for proteasomal degradation, induced the expression of both Hif-1α and Hif-2α (Figure 1b), indicating that the effects of PTH are specific for Hif-1α. PTH administration also increased Hif-1α expression in vivo as PTH treated mice (40 µg·kg−1) exhibited robust expression of Hif-1α in osteoblasts lining trabecular bone surfaces, a subset of osteocytes, and marrow components (Figure 1c), while saline treated animals exhibited only weak expression in these cell populations. Both in vitro (Figure 1d) and in vivo (Figure 1e), the levels of Hif-1α mRNA were unaffected by PTH stimulation, suggesting that the induction of Hif-1α protein occurs via a post-transcriptional mechanism.

Parathyroid hormone increases the expression of Hif-1α both in vitro and in vivo. (a and b) Levels of Hif-1α, Hif-2α and Hif-1β protein were measured in wild-type osteoblasts at the indicated time points after treatment with 10 nmol·L−1 PTH (a) and after 6 h of treatment with 200 nmol·L−1 DFO (b). (c) Immunohistochemical analysis of Hif-1α expression in the trabecular bone of 10-week-old female wild-type mice treated with 40 μg·kg−1 PTH or vehicle for 4 h. Representative tissue sections show increased Hif-1α expression in the osteoblasts (arrows) of PTH-treated mice relative to untreated controls. (d and e) mRNA levels of Hif-1α and Hif-2α were measured in vitro (d) and in vivo (e) after treatment with PTH or vehicle. Spp1 (Osteopontin) and Ptgs2 (Cox2) were used as controls. *P≤0.05. DFO, desferoxamine.
cAMP/protein kinase A (PKA) signaling induces Hif-1α expression in response to PTH
PTH could increase Hif-1α protein levels without affecting Hif-1α transcription by specifically enhancing Hif-1α translation or by inhibiting proteasomal degradation. Since PTH retained the capacity to increase Hif-1α protein levels in osteoblasts deficient for Vhl (data not shown), we focused on the induction of new Hif-1α synthesis. As expected, pre-treatment of osteoblast cultures with cycloheximide, to inhibit new protein synthesis, abolished the effect of PTH on Hif-1α protein levels (Figure 2a and 2d). Moreover, increases in the phosphorylation of Akt (S473) and p70 S6 kinase (Figure 2b) indicated that PTH activates mTOR, a key regulator of Hif expression in response to anabolic signals.39,40 Adenoviral Cre-mediated disruption of mTOR expression in osteoblasts containing mTORflox/flox alleles, via an 83% reduction in mTOR mRNA levels, completely inhibited the effect of PTH on Hif-1α protein (Figure 2c and 2d).

cAMP/PKA signaling induces Hif-1α expression. (a) Levels of Hif-1α protein were measured in wild-type osteoblasts following 4 h of treatment with 10 nmol·L−1 PTH or vehicle (NT), preceded by 30 min of treatment with 100 μg·mL−1 cycloheximide (Chx). (b) Levels of Akt (S473) and p70 S6 kinase protein phosphorylation were measured in wild-type osteoblasts following 1 h of treatment with 10 nmol·L−1 PTH. (c) Hif-1α protein expression was measured in control and mTOR-deficient osteoblasts following treatment with 10 nmol·L−1 PTH for 4 h. (d) Densitometric quantitation of Hif-1α protein as shown in a and c. (e and g) Akt and p70 S6 kinase phosphorylation (e) and Hif-1α protein expression (g) were measured in wild-type osteoblasts following 30 min (e) or 6 hours (g) of treatment with Forskolin at the indicated concentrations. (f) Quanitification of Akt phosphorylation shown in e in Forskolin-treated osteoblasts relative to untreated. (h) Relative expression of Hif-1α protein as shown in g. (i and l) Levels of Akt and p70 S6 kinase phosphorylation (i) and Hif-1α protein expression (l) were measured following 30 min (i) or 6 h (l) of treatment with 10 nmol·L−1 PTH, preceded by 30 min of treatment with 10 μmol·L−1 H-89. (j and k) Quantification of Akt (j) and p70 S6 kinase (k) phosphorylation shown in i in PTH- and/or H-89-treated osteoblasts relative to untreated. (m) Relative expression of Hif-1α protein as shown in l. *P≤0.05.
We next explored the signaling mechanisms by which PTH activates mTOR and ultimately increases Hif-1α protein. Because an increase in cellular cAMP signaling is a primary response to PTH binding to its receptor,6,12 we initially assessed the effect of pharmacologically raising cAMP levels on mTOR activity and Hif-1α expression. Forskolin, which activates adenylyl cyclase, dose-dependently stimulated the phosphorylation of Akt and p70 S6 kinase (Figure 2e and 2f), indicating that mTOR was activated, and increased the levels of Hif-1α protein (Figure 2g and 2h). To confirm these results, we pre-treated osteoblast cultures with H-89 to antagonize the activity of PKA, the downstream mediator of cAMP signaling. This approach greatly impaired the ability of PTH to stimulate the phosphorylation of Akt and p70 S6 kinase (Figure 2i–2k), even though baseline levels of p70 phosphorylation were increased by H-89, and abolished the increase in Hif-1α protein (Figure 2l and 2m). Together, these data suggest a mechanism whereby PTH activates cAMP/PKA signaling which in turn activates the mTOR pathway to regulate Hif-1α expression.
ΔHif-1α mice are more sensitive to anabolic PTH treatment
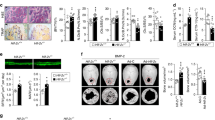
To directly assess the effect of Hif-1α expression on the anabolic response of bone to PTH, we generated cohorts of 10-week-old female control and ΔHif-1α mice and administered daily injections of PTH or saline, as a control, for 6 weeks. MicroCT analysis revealed equivalent trabecular bone architecture in the distal femur of saline treated control and ΔHif-1α mice, which is consistent with our previous study27 that demonstrated a normalization of bone volume in Hif-1α mutants (Figure 3a–d). PTH injections (40 µg·kg−1 BW, Body weight) produced the predicted anabolic response and increased trabecular bone volume by 155.41% in control mice (Figure 3a and 3b) by increasing trabecular number (Figure 3c) and thickness (Figure 3d). In Hif-1α mutants, PTH increased bone volume an additional 22.86% relative to the treated control mice (182.19% increase versus ΔHif-1α, saline-treated controls), due to significantly larger increases in trabecular thickness and slightly larger increases in trabecular number. PTH also significantly increased cortical bone thickness, but the effect was indistinguishable between control and Hif-1α mutant mice (data not shown). Likewise, increases in body weight were equivalent among control and Hif-1α mutant mice (data not shown). When we reduced the daily dose of PTH to 20 µg·kg−1, the increase in trabecular bone volume in the mutant mice (182.79% increase versus ΔHif-1α, saline-treated controls) was identical to that of the 40 µg·kg−1 treatment group, while the percent increase in the control mice was reduced by 29.91% (Figure 3a and 3b). Taken together, these data suggest that disrupting the expression of Hif-1α increases the sensitivity of bone to intermittent PTH.

Bone formation after PTH administration is increased in mice lacking Hif-1α in osteoblasts and osteocytes. Ten-week-old female control and Hif-1α mutant (ΔHif-1α) mice were treated with vehicle or PTH for 6 weeks by daily s.c. injection. (a) Representative microCT images illustrate trabecular bone structure in the distal femur of control and ΔHif-1α mice after treatment with PTH at the indicated doses. (b–d) Bone volume per tissue volume (BV/TV) (b), trabecular number (Tb. N) (c) and trabecular thickness (Tb. Th) (d) were quantified by microCT. (e–g) The mineralizing surface per bone surface (MS/BS) (e), mineral apposition rate (MAR) (f) and bone formation rate per bone surface (BFR/BS) (g) were assessed by dynamic histomorphometry. (h and i) Levels of P1NP (h) and CTx (i) were measured in serum collected from control and ΔHif-1α mice. Results from e through i are presented as % change from baseline levels in saline-treated animals, which were equivalent in control and ΔHif-1α mice. *P≤0.05 vs. control mice with matched treatment. P1NP, amino pro-peptide of type 1 collagen; CTx, C-terminal telopeptide; s.c., subcutaneous.
To understand the cellular basis for the enhanced anabolic response in ΔHif-1α mice, we performed dynamic histomorphometric and serological analyses of bone formation. Since all measures were equivalent in the saline-treated control and ΔHif-1α mice, results are presented as the relative increase above this baseline level (Figure 3e–3i). PTH increased the mineralizing surface per bone surface to a similar extent in control and ΔHif-1α mice (Figure 3e), but the mutant mice exhibited a 23.2% increase in mineral apposition rate while no effect of PTH on this parameter was observed in the control mice (Figure 3f). This led to a greater overall increase in bone formation rate in the mutant mice relative to the control mice (Figure 3g). Similarly, serum levels of P1NP (Figure 3h), a marker of bone formation, were increased to a greater extent in the mutant mice relative to controls, while the levels of C-terminal telopeptide (Figure 3i), a marker of bone resorption, were increased to a similar degree. Thus, the amplified sensitivity of Hif-1α mutant mice to PTH and augmented bone formation response appear to be due to an increase in the functional activity of individual osteoblasts.
Hif-1α antagonizes the actions of β-catenin after PTH stimulation
Finally, we assessed cellular signaling mechanisms that might account for the increased responsiveness of Hif-1α mutant mice to PTH. Calvarial osteoblasts were isolated from Hif-1αflox/flox mice and infected with adenoviral constructs expressing Cre to eliminate Hif-1α expression (Figure 4a) or green fluorescent protein as a control. We next examined the effect of eliminating Hif-1α expression on the activation of the primary PTH-responsive pathways, but the increase in cellular cAMP levels (Figure 4b) and the phosphorylation of Creb and Erk (Figure 4c) were similar in control and Hif-1α deficient osteoblasts. By contrast, the increase in Axin2 and Nkd2 mRNA levels was enhanced after PTH treatment in ΔHif-1α osteoblasts relative to those of controls (Figure 4d and 4e), suggesting that the activation of β-catenin was increased.41 To test the specificity of this apparent inhibitory effect of Hif-1α on β-catenin activity, we overexpressed Hif-1α by eliminating the expression of Vhl (Figure 4f), and as expected this genetic manipulation impaired the ability of PTH to increase the expression of Axin2 (Figure 4g). The expression of Hif-1α in response to PTH did not alter the accumulation or nuclear localization of β-catenin (Figure 4h), but rather Hif-1α directly interacted with β-catenin (Figure 4i) and acted to inhibit the binding of β-catenin to the promoter of target genes (Figure 4j). We observed a similar effect in vivo as PTH produced a significant increase in Axin2 mRNA levels in the femurs of ΔHif-1α mice, but not those of control mice. These data imply that the enhanced bone formation response evident in Hif-1α mutant mice stems from the elimination of Hif-1α-mediated suppression of β-catenin signaling.

Hif-1α inhibits β-catenin activity following PTH administration.(a) Relative expression of Hif-1α mRNA in osteoblasts following adenoviral Cre-mediated deletion. (b) Levels of cAMP production were measured by immunoassay after control and Hif-1α-deficient (ΔHif-1α) osteoblasts were treated with 10 nmol·L−1 PTH for 15 min. (c) Phosphorylation levels of Creb and Erk in control and ΔHif-1α osteoblasts were measured following 0–30 min of treatment with 10 nmol·L−1 PTH. (d and e) Relative expression of Axin2 (d) and Nkd2 (e) mRNA in control and ΔHif-1α osteoblasts treated with 10 nmol·L−1 PTH for 8 h or left untreated (NT). (f) Immunoblot analysis of Hif-1α levels in control and Vhl-deficient (ΔVhl) osteoblasts 48 h after adenoviral Cre infection. (g) Relative expression of Axin2 mRNA in control and ΔVhl osteoblasts treated with 10 nmol·L−1 PTH for 4 h or left untreated (NT). (h) Protein levels of β-catenin in nuclear extracts isolated from control and ΔHif-1α osteoblasts treated with 10 nmol·L−1 PTH for 4 h or left untreated (NT). Tbp was used as a nuclear control. (i) IP analysis of Hif-1α and β-catenin interaction following 4 h of 10 nmol·L−1 PTH treatment in wild-type cells. (j) Quantitative chromatin IP analysis of β-catenin occupancy at the Axin2 promoter in control and ΔHif-1α osteoblasts after 8 h of treatment with 10 nmol·L−1 PTH. (k) Relative expression of Axin2 mRNA in samples collected from the femur of control and ΔHif-1α mice after treatment with saline (NT) or 40 µg·kg−1 PTH for 16 h. *P≤0.05. IP, immunoprecipitation; Tbp, TATA-binding protein.
Discussion
In this study, we demonstrate that the transcription factor Hif-1α acts to suppress the anabolic actions of parathyroid hormone. Hif-1α protein levels were rapidly upregulated both in vitro and in vivo by PTH stimulation and mice rendered deficient for Hif-1α in osteoblasts and osteocytes were more responsive to intermittent administration of the hormone. The more dramatic increase in bone formation evident in Hif-1α mutant mice appears to result from an increase in the performance of individual osteoblasts secondary to an enhancement of β-catenin target gene expression.
In addition to hypoxia, Hif-1α is induced by a number of anabolic signals relevant to bone metabolism. Mechanical loading,42 growth factors and paracrine factors like prostaglandins43–45 all increase Hif-1α protein levels. Some of these factors have also been implicated in the anabolic response of bone to PTH. Insulin-like growth factor-1, for instance, induces Hif-1α expression,46 and removal of its receptor diminishes bone formation in response to the hormone.13 While insulin-like growth factor-1 might partially contribute to the induction of Hif-1α after PTH stimulation, the rapid effect we observed suggests a more direct effect on Hif-1α synthesis. Indeed, our data suggest that PTH activates cAMP/PKA signaling to increase the activity of mTOR, which can directly regulate Hif-1α translation.39,40,47 While not directly examined here, studies in other tissues indicate that PKA can activate mTOR signaling via a number of mechanisms, including the engagement of PI3K/Akt signaling and the phosphorylation of TOR complex components.48,49
As alluded to above, mTOR signaling has also been demonstrated to be an important component of the anabolic response of bone to PTH, as rapamycin treatment inhibits the increase in trabecular bone volume resulting from high-dose PTH administration by reducing osteoblastic activity.29 In light of our data, it would appear that mTOR plays a dual role in the response of bone to intermittent PTH, facilitating anabolism while also establishing a mechanism to suppress anabolic signaling via induction of Hif-1α expression. We focused our analysis on the effects of Hif-1α on β-catenin signaling because of its well-documented anabolic role in the skeleton, but we cannot rule out the possibility that Hif-1α also regulates the expression of factors that inhibit mTOR signaling.50
Even though angiogenesis is a well-explored response to increased Hif-1α signaling and PTH has been shown to induce skeletal vascular remodeling,30 it does not appear that this effect factors into the anabolic response we observed. If vascular remodeling and the relocalization of blood vessels to sites of new bone formation were regulated by Hif-1α, we would have expected the disruption of Hif-1α expression to impair the osteo-anabolic response. However, we cannot completely rule out a compensatory effect of the closely related Hif-2α. Rather, as we have suggested previously,27 it is likely that Hif-1α assumes an inhibitory role in the mature skeleton to prevent unchecked anabolic signaling and the generation of signals that impinge on cellular function. Hif-1α is induced by reactive oxygen species, and Hif-1-generated signals in turn reduce new oxidant production.51–54 Therefore, induction of Hif-1α may ensure cellular longevity and facilitate cellular repair after the generation of cellular stressors during an anabolic response.
The results presented here are consistent with our previous finding that Hif-1α acts to inhibit the anabolic response to a tibia-loading regime and potentially does so by suppressing β-catenin activity.27 However, several important differences exist when the cellular basis of each response is considered. Our mechanical loading study demonstrated that Hif-1α inhibited the anabolic response in the cortical bone envelope, but in these studies, PTH produced similar increases in cortical bone thickness in control and mutant mice. Likewise, the number of osteoblasts activated by mechanical loading was greatly increased in Hif-1α mutant mice, but the effect of PTH on bone formation in these mice appears to be related to a larger increase in the functional output of individual osteoblasts. Here, the mineralizing surface was similarly enhanced in control and mutant mice, but the mineral apposition rate and P1NP levels were enhanced by the genetic removal of Hif-1α. While we cannot exclude the possibility that the differential effects are simply the result of different bone compartments, these data suggest that the suppressive actions of Hif-1α may be dependent on the cellular context and the stimulus.
Nonetheless, the significant increase in the anabolic effect of each of these stimuli suggests that Hif-1α activity could be targeted in therapeutic paradigms. Previous studies have identified pharmacological molecules that impair Hif-1α transcriptional activity or interaction with binding partners.55–57 While these studies have primarily focused on the prevention of tumor growth and tumor-induced angiogenesis, these agents or molecules with similar functions could be adapted to enhance anabolic therapies in bone. After prolonged treatment with intermittent PTH, markers of bone formation begin to decline, suggestive of the development of a resistance to the anabolic effects of the therapy.58,59 Whether increased expression of Hif-1α contributes to this effect will require additional studies. However, our studies suggest that an agent that inhibits the expression of Hif-1α or impairs the interaction of Hif-1α and β-catenin could be used to lower the therapeutic dose of PTH necessary to decrease fracture risk or increase the anabolic response.
In summary, our studies support a role for Hif-1α as a negative regulator of osteo-anabolic signaling. In early development, Hif-1α functions in bone cells to facilitate the vascularization of long bones, a process that is required for normal bone acquisition. As bone matures, Hif-1α assumes a new function that likely acts to restrain osteoblast and osteocyte activity and does so by interfering with a key component of the Wnt signaling pathway. Our studies provide a broader understanding of the molecular physiology of Hif-1α in bone cells and may lead to the design of strategies to augment skeletal therapeutics.
References
Dempster DW, Cosman F, Parisien M, Shen V, Lindsay R . Anabolic actions of parathyroid hormone on bone. Endocr Rev 1993; 14: 690–709.
Finkelstein JS, Klibanski A, Schaefer EH, Hornstein MD, Schiff I, Neer RM . Parathyroid hormone for the prevention of bone loss induced by estrogen deficiency. N Engl J Med 1994; 331: 1618–1623.
Hansen S, Hauge EM, Beck Jensen JE, Brixen K . Differing effects of PTH 1–34, PTH 1–84, and zoledronic acid on bone microarchitecture and estimated strength in postmenopausal women with osteoporosis: an 18-month open-labeled observational study using HR-pQCT. J Bone Miner Res 2013; 28: 736–745.
Kurland ES, Cosman F, McMahon DJ, Rosen CJ, Lindsay R, Bilezikian JP . Parathyroid hormone as a therapy for idiopathic osteoporosis in men: effects on bone mineral density and bone markers. J Clin Endocrinol Metab 2000; 85: 3069–3076.
Jilka RL . Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone 2007; 40: 1434–1446.
Abou-Samra AB, Juppner H, Force T et al. Expression cloning of a common receptor for parathyroid hormone and parathyroid hormone-related peptide from rat osteoblast-like cells: a single receptor stimulates intracellular accumulation of both cAMP and inositol trisphosphates and increases intracellular free calcium. Proc Natl Acad Sci USA 1992; 89: 2732–2736.
Iida-Klein A, Zhou H, Lu SS et al. Anabolic action of parathyroid hormone is skeletal site specific at the tissue and cellular levels in mice. J Bone Miner Res 2002; 17: 808–816.
Dobnig H, Turner RT . The effects of programmed administration of human parathyroid hormone fragment (1–34) on bone histomorphometry and serum chemistry in rats. Endocrinology 1997; 138: 4607–4612.
Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC . Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest 1999; 104: 439–446.
Wu X, Pang L, Lei W et al. Inhibition of Sca-1-positive skeletal stem cell recruitment by alendronate blunts the anabolic effects of parathyroid hormone on bone remodeling. Cell Stem Cell 2010; 7: 571–580.
Dobnig H, Turner RT . Evidence that intermittent treatment with parathyroid hormone increases bone formation in adult rats by activation of bone lining cells. Endocrinology 1995; 136: 3632–3638.
Rodan SB, Rodan GA . The effect of parathyroid hormone and thyrocalcitonin on the accumulation of cyclic adenosine 3′:5′-monophosphate in freshly isolated bone cells. J Biol Chem 1974; 249: 3068–3074.
Wang Y, Nishida S, Boudignon BM et al. IGF-I receptor is required for the anabolic actions of parathyroid hormone on bone. J Bone Miner Res 2007; 22: 1329–1337.
Yakar S, Bouxsein ML, Canalis E et al. The ternary IGF complex influences postnatal bone acquisition and the skeletal response to intermittent parathyroid hormone. J Endocrinol 2006; 189: 289–299.
Yamaguchi M, Ogata N, Shinoda Y et al. Insulin receptor substrate-1 is required for bone anabolic function of parathyroid hormone in mice. Endocrinology 2005; 146: 2620–2628.
Qiu T, Wu X, Zhang F, Clemens TL, Wan M, Cao X . TGF-beta type II receptor phosphorylates PTH receptor to integrate bone remodelling signalling. Nat Cell Biol 2010; 12: 224–234.
Li C, Xing Q, Yu B et al. Disruption of LRP6 in osteoblasts blunts the bone anabolic activity of PTH. J Bone Miner Res 2013; 28: 2094–2108.
Robling AG, Kedlaya R, Ellis SN et al. Anabolic and catabolic regimens of human parathyroid hormone 1–34 elicit bone- and envelope-specific attenuation of skeletal effects in Sost-deficient mice. Endocrinology 2011; 152: 2963–2975.
Robling AG, Childress P, Yu J et al. Nmp4/CIZ suppresses parathyroid hormone-induced increases in trabecular bone. J Cell Physiol 2009; 219: 734–743.
Childress P, Philip BK, Robling AG et al. Nmp4/CIZ suppresses the response of bone to anabolic parathyroid hormone by regulating both osteoblasts and osteoclasts. Calcif Tissue Int 2011; 89: 74–89.
Semenza GL . Hypoxia-inducible factors in physiology and medicine. Cell 2012; 148: 399–408.
Semenza GL . HIF-1, O2, and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell 2001; 107: 1–3.
Mole DR, Blancher C, Copley RR et al. Genome-wide association of hypoxia-inducible factor (HIF)-1α and HIF-2α DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem 2009; 284: 16767–16775.
Kulshreshtha R, Ferracin M, Wojcik SE et al. A microRNA signature of hypoxia. Mol Cell Biol 2007; 27: 1859–1867.
Shomento SH, Wan C, Cao X et al. Hypoxia-inducible factors 1α and 2α exert both distinct and overlapping functions in long bone development. J Cell Biochem 2010; 109: 196–204.
Wang Y, Wan C, Deng L et al. The hypoxia-inducible factor α pathway couples angiogenesis to osteogenesis during skeletal development. J Clin Invest 2007; 117: 1616–1626.
Riddle RC, Leslie JM, Gross TS, Clemens TL . Hypoxia-inducible factor-1α protein negatively regulates load-induced bone formation. J Biol Chem 2011; 286: 44449–44456.
Akeno N, Robins J, Zhang M, Czyzyk-Krzeska MF, Clemens TL . Induction of vascular endothelial growth factor by IGF-I in osteoblast-like cells is mediated by the PI3K signaling pathway through the hypoxia-inducible factor-2α. Endocrinology 2002; 143: 420–425.
Niziolek PJ, Murthy S, Ellis SN et al. Rapamycin impairs trabecular bone acquisition from high-dose but not low-dose intermittent parathyroid hormone treatment. J Cell Physiol 2009; 221: 579–585.
Prisby R, Guignandon A, Vanden-Bossche A et al. Intermittent PTH(1–84) is osteoanabolic but not osteoangiogenic and relocates bone marrow blood vessels closer to bone-forming sites. J Bone Miner Res 2011; 26: 2583–2596.
Zhang M, Xuan S, Bouxsein ML et al., Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem 2002; 277: 44005–44012.
Ryan HE, Lo J, Johnson RS . HIF-1α is required for solid tumor formation and embryonic vascularization. EMBO J 1998; 17: 3005–3015.
Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, Simon MC . Acute postnatal ablation of Hif-2α results in anemia. Proc Natl Acad Sci USA 2007; 104: 2301–2306.
Haase VH, Glickman JN, Socolovsky M, Jaenisch R . Vascular tumors in livers with targeted inactivation of the von Hippel–Lindau tumor suppressor. Proc Natl Acad Sci USA 2001; 98: 1583–1588.
Lang CH, Frost RA, Bronson SK, Lynch CJ, Vary TC . Skeletal muscle protein balance in mTOR heterozygous mice in response to inflammation and leucine. Am J Physiol Endocrinol Metab 2010; 298: E1283–E1294.
Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R . Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res 2010; 25: 1468–1486.
Dempster DW, Compston JE, Drezner MK et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 2013; 28: 2–17.
Parfitt AM, Drezner MK, Glorieux FH et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 1987; 2: 595–610.
Land SC, Tee AR . Hypoxia-inducible factor 1α is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem 2007; 282: 20534–20543.
Hudson CC, Liu M, Chiang GG et al. Regulation of hypoxia-inducible factor 1α expression and function by the mammalian target of rapamycin. Mol Cell Biol 2002; 22: 7004–7014.
Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F . Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol 2002; 22: 1172–1183.
Milkiewicz M, Doyle JL, Fudalewski T, Ispanovic E, Aghasi M, Haas TL . HIF-1α and HIF-2α play a central role in stretch-induced but not shear-stress-induced angiogenesis in rat skeletal muscle. J Physiol 2007; 583: 753–766.
Liu XH, Kirschenbaum A, Lu M et al. Prostaglandin E2 induces hypoxia-inducible factor-1α stabilization and nuclear localization in a human prostate cancer cell line. J Biol Chem 2002; 277: 50081–50086.
Semenza G . Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol 2002; 64: 993–998.
Semenza GL . HIF-1 and human disease: one highly involved factor. Genes Dev 2000; 14: 1983–1991.
Feldser D, Agani F, Iyer NV, Pak B, Ferreira G, Semenza GL . Reciprocal positive regulation of hypoxia-inducible factor 1α and insulin-like growth factor 2. Cancer Res 1999; 59: 3915–3918.
Yamashita R, Suzuki Y, Takeuchi N et al. Comprehensive detection of human terminal oligo-pyrimidine (TOP) genes and analysis of their characteristics. Nucleic Acids Res 2008; 36: 3707–3715.
Blancquaert S, Wang L, Paternot S et al. cAMP-dependent activation of mammalian target of rapamycin (mTOR) in thyroid cells. Implication in mitogenesis and activation of CDK4. Mol Endocrinol 2010; 24: 1453–1468.
Alam H, Maizels ET, Park Y et al. Follicle-stimulating hormone activation of hypoxia-inducible factor-1 by the phosphatidylinositol 3-kinase/AKT/Ras homolog enriched in brain (Rheb)/mammalian target of rapamycin (mTOR) pathway is necessary for induction of select protein markers of follicular differentiation. J Biol Chem 2004; 279: 19431–19440.
Brugarolas J, Lei K, Hurley RL et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 2004; 18: 2893–2904.
Simon MC . Mitochondrial reactive oxygen species are required for hypoxic HIF α stabilization. Adv Exp Med Biol 2006; 588: 165–170.
Brunelle JK, Bell EL, Quesada NM et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab 2005; 1: 409–414.
Kim JW, Tchernyshyov I, Semenza GL, Dang CV . HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006; 3: 177–185.
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC . HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 2006; 3: 187–197.
Wong CC, Zhang H, Gilkes DM et al. Inhibitors of hypoxia-inducible factor 1 block breast cancer metastatic niche formation and lung metastasis. J Mol Med (Berl) 2012; 90: 803–815.
Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL . Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci USA 2009; 106: 17910–17915.
Zhang H, Qian DZ, Tan YS et al. Digoxin and other cardiac glycosides inhibit HIF-1α synthesis and block tumor growth. Proc Natl Acad Sci USA 2008; 105: 19579–19586.
Girotra M, Rubin MR, Bilezikian JP . The use of parathyroid hormone in the treatment of osteoporosis. Rev Endocr Metab Disord 2006; 7: 113–121.
Rubin MR, Bilezikian JP . The anabolic effects of parathyroid hormone therapy. Clin Geriatr Med 2003; 19: 415–432.
Acknowledgements
We thank Dr C Lynch for providing the mTORflox/flox mice. Support was provided by a Career Development Award (RCR, BX001284) from the Veterans Administration.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Frey, J., Stonko, D., Faugere, MC. et al. Hypoxia-inducible factor-1α restricts the anabolic actions of parathyroid hormone. Bone Res 2, 14005 (2014). https://doi.org/10.1038/boneres.2014.5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/boneres.2014.5
This article is cited by
-
Hypoxia-inducible factor signaling in vascular calcification in chronic kidney disease patients
Journal of Nephrology (2022)
-
Oxygen desaturation during the 6-min walk test as a risk for osteoporosis in non-cystic fibrosis bronchiectasis
BMC Pulmonary Medicine (2019)
-
Hypoxia Signaling in the Skeleton: Implications for Bone Health
Current Osteoporosis Reports (2019)
