
Abstract
Background:
Copanlisib is a pan-class I phosphatidylinositol 3-kinase (PI3K) inhibitor with predominant PI3K-α/δ activity that has demonstrated clinical activity and manageable safety when administered as monotherapy in a phase II study. Combination therapy may overcome compensatory signalling that could occur with PI3K pathway inhibition, resulting in enhanced inhibitory activity, and preclinical studies of copanlisib with gemcitabine have demonstrated potent anti-tumour activity in vivo.
Methods:
A phase I, open-label, dose-escalation study to evaluate the safety, tolerability and recommended phase II dose (RP2D) of copanlisib with gemcitabine or with cisplatin plus gemcitabine (CisGem) in patients with advanced malignancies, including an expansion cohort in patients with biliary tract cancer (BTC) at the RP2D of copanlisib plus CisGem. Copanlisib and gemcitabine were administered on days 1, 8 and 15 of a 28-day cycle; maximum tolerated dose (MTD) and RP2D of copanlisib were determined. Copanlisib plus CisGem was administered on days 1 and 8 of a 21-day cycle; pharmacokinetics and biomarkers were assessed.
Results:
Fifty patients received treatment as follows: dose-escalation cohorts, n=16; copanlisib plus CisGem cohort, n=14; and BTC expansion cohort, n=20. Copanlisib 0.8 mg kg−1 plus gemcitabine was the MTD and RP2D for both combinations. Common treatment-emergent adverse events included nausea (86%), hyperglycaemia (80%) and decreased platelet count (80%). Copanlisib exposure displayed a dose-proportional increase. No differences were observed upon co-administration of CisGem. Response rates were as follows: copanlisib plus gemcitabine, 6.3% (one partial response in a patient with peritoneal carcinoma); copanlisib plus CisGem, 12% (one complete response and three partial responses all in patients with BTC (response rate 17.4% in patients with BTC)). Mutations were detected in PIK3CA (1 out of 43), KRAS (10 out of 43) and BRAF (2 out of 22), with phosphate and tensin homologue protein loss in 41% (12 out of 29).
Conclusions:
Copanlisib plus CisGem demonstrated a manageable safety profile, favourable pharmacokinetics, and potentially promising clinical response.
Similar content being viewed by others

Main
Cellular metabolism, growth and differentiation are dependent on signalling through the phosphatidylinositol 3-kinase (PI3K)/α-serine/threonine-protein kinase (AKT)/mammalian target of rapamycin (mTOR) pathway, and dysregulation of this pathway has been shown to drive tumourigenesis (Yuan and Cantley, 2008; Ihle and Powis, 2009). Upregulation of the PI3K/AKT/mTOR pathway attributed to mutations in PIK3CA has been identified in many cancer types, including biliary tract cancer (BTC) (Deshpande et al, 2011; Singh et al, 2015; Li et al, 2016).
Tumour suppressor gene phosphate and tensin homologue (PTEN) is involved in cell cycle regulation and is mutated in many cancers. PTEN mutations lead to activation of the PI3K/AKT pathway, and loss of PTEN function results in increased phosphatidylinositol (3,4,5)-trisphosphate levels and subsequent AKT phosphorylation and modulation of its downstream molecular oncogenic process (Wang et al, 2015). The PI3K/AKT/mTOR pathway is implicated in chemotherapy resistance (Lee et al, 2015). Overcoming this tumour survival mechanism by blocking intracellular signalling through the PI3K/AKT/mTOR pathway is a potential therapeutic target.
Copanlisib (Bayer AG, Leverkusen, Germany) is an intravenous, potent, highly selective and reversible pan-class I PI3K inhibitor with predominant activity against PI3K-δ and PI3K-α isoforms (Liu et al, 2013; Haike et al, 2014). In preclinical studies, copanlisib demonstrated anti-tumour activity in PIK3CA-mutated cells (Liu et al, 2013). The first-in-human study of copanlisib monotherapy determined the maximum tolerated dose (MTD) to be 0.8 mg kg−1, with promising efficacy in patients with solid tumours and haematological malignancies (Patnaik et al, 2016), including clinically meaningful responses in patients with relapsed or refractory indolent or aggressive malignant lymphoma (Dreyling et al, 2017a, b).
Inhibition of the PI3K/AKT/mTOR pathway releases negative feedback that can result in activation of compensatory signalling pathways; therefore, combination therapy regimens have been suggested to overcome this (Lee et al, 2015).
Cisplatin and gemcitabine are the current standard of care in many advanced cancer types, including pancreatic, bladder and BTC (cholangiocarcinoma and gallbladder cancer). Most BTCs are advanced or metastatic at diagnosis and median overall survival is typically <1 year (Ebata et al, 2017); therefore, combining DNA-targeting therapies with copanlisib may be an attractive treatment strategy. In preclinical studies, gemcitabine combined with copanlisib demonstrated anti-tumour activity in a mutant BTC model in nude mice (Hägebarth et al, 2012).
Here we report the results of a phase I study to determine the safety, tolerability and recommended phase II dose (RP2D) of copanlisib in combination with gemcitabine or with cisplatin plus gemcitabine (CisGem) in patients with advanced solid malignancies, including BTC (www.clinicaltrials.gov, Identifier: NCT01460537).
Methods
Study design
This was a phase I, multicentre, open-label, non-randomised, dose-escalation study. Patients were initially enrolled for treatment with copanlisib plus gemcitabine alone to evaluate safety and tolerability. Intravenous gemcitabine (1000 mg m−2) and copanlisib (1 h) at a starting dose of 0.6 mg kg−1 were administered on days 1, 8 and 15 of a 28-day treatment cycle. Copanlisib dose escalation was performed following a standard 3+3 design, planned to a maximum dose of 0.8 mg kg−1 in a second cohort. Details of dose-limiting toxicities (DLTs) and permitted dose reductions are provided in the Supplementary Materials section, available online at British Journal of Cancer.
Once the MTD was determined for copanlisib with gemcitabine, this copanlisib dose was administered in combination with intravenous CisGem at fixed doses of 25 mg m−2 and 1000 mg m−2, respectively, on days 1 and 8 of a 21-day treatment cycle. If gemcitabine (or cisplatin) was discontinued for toxicity, copanlisib could be continued as monotherapy or in a doublet at the discretion of the investigator if a clinical benefit was observed.
Upon determination of the RP2D of copanlisib plus CisGem, enrolment in an expansion cohort of up to 20 patients with BTC was planned.
Study population
Patients were eligible if they were aged ⩾18 years with histologically or cytologically confirmed, advanced or refractory solid tumours, and if gemcitabine and cisplatin were medically appropriate. Patients were eligible for the expansion cohort if they had a histologically confirmed diagnosis of BTC (including intrahepatic cholangiocarcinoma, extrahepatic cholangiocarcinoma or gallbladder cancer). Patients were required to have at least one measurable lesion or evaluable disease, as determined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, and an Eastern Cooperative Oncology Group performance status of 0 or 1. Further inclusion and exclusion criteria are included in the Supplementary Methods section.
This study was approved by independent ethics committees and institutional review boards for each study site and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. All patients provided written, informed consent before participation.
Objectives and assessments
The primary objectives were to determine the safety, tolerability and RP2D of copanlisib plus gemcitabine and of copanlisib plus CisGem, and to characterise the pharmacokinetics (PK) of copanlisib, gemcitabine and cisplatin when administered concomitantly in patients with advanced malignancies. Secondary objectives included assessment of clinical response and biomarkers that may predict response to the combination drugs.
Adverse events (AEs) and serious AEs were observed from enrolment until 30 days after the last dose of study treatment. Laboratory toxicities and AEs were graded by the National Cancer Institute’s Common Terminology Criteria for Adverse Events version 4.0.
Tumour assessment by computed tomography scan or magnetic resonance imaging was performed at screening and within 7 days of the end of every second cycle using RECIST version 1.1.
PIK3CA and KRAS mutations were assayed in circulating tumour DNA isolated from pre-treatment plasma samples (collected during the screening period) using beads, emulsions, amplification and magnetics technology (Sysmex Inostics GmbH, Hamburg, Germany) (Diehl et al, 2006). Next-generation sequencing of DNA isolated from formalin-fixed, paraffin-embedded pre-treatment tumour samples (either archival or at screening) was performed using the FoundationOne next-generation sequencing panel (Foundation Medicine, Cambridge, MA, USA). Tumour PTEN protein levels were analysed by immunohistochemistry in pre-treatment tumour tissue samples.
Further details of assessments and PK evaluation are provided in the Supplementary Methods section.
Statistical analyses
Pharmacokinetic concentration calculated for each of the sampling time points included arithmetic mean, standard deviation and coefficient of variation, geometric mean, geometric standard deviation and coefficient of variation, minimum, median, maximum value and the number of measurements. Means at any time were calculated only if at least two out of three of the individual data were measured and were above the lowest limit of quantification (LLOQ). For the calculation of the mean value, a data point below the LLOQ was substituted by one-half of this limit. PK characteristics, except for time to reach maximum plasma concentration after single-dose administration (tmax) and time of last observed plasma concentration value above the LLOQ (tlast), were summarised using the statistics mentioned previously. Tmax and tlast were described using minimum, maximum, median and frequency counts.
Pharmacokinetic parameters of primary interest for copanlisib, copanlisib metabolite M-1, gemcitabine, gemcitabine metabolite 2’,2’-difluorodeoxyuridine and total and free platinum were maximum observed plasma concentration after single-dose administration (Cmax) and area under the plasma concentration vs time curve (AUC) from time 0 to the last observed plasma concentration value above the LLOQ (AUC(0–tlast)). The metabolite ratio of copanlisib metabolite M-1 to copanlisib was calculated for AUC(0–tlast) and AUC from 0 to 25 h after the start of the infusion (AUC(0–25)) using molar concentrations. Dose-normalised PK characteristics were calculated for Cmax, AUC(0–tlast) and AUC(0–25).
For immunohistochemistry results, H-score values were calculated to determine the average intensity of positive staining given weight by the percentage of cells showing positive staining (3 × (% of tumour cells staining positive at intensity 3+)+2 × (% of tumour cells staining positive at intensity 2+)+1 × (% of tumour cells staining positive at intensity 1+)).
Results
Baseline patient demographics and disease characteristics
Fifty patients were assigned to treatment, a small majority of whom were female (56%). Median age at screening was 63 years (range 33–79) (Table 1). Most patients had intrahepatic bile duct cancer (16 patients, 32%) and gallbladder cancer (seven patients, 14%), primarily because of the BTC expansion cohort (23 patients in total). The majority (39 patients, 78%) had received at least one prior systemic anti-cancer therapy (median number of regimens, 3 (range 1–11)); 11 patients with BTC had not received any prior systemic anti-cancer therapy.
Sixteen patients were treated in the copanlisib plus gemcitabine dose-escalation cohorts; eight patients received the starting dose of 0.6 mg kg−1 and eight patients received the maximum dose of 0.8 mg kg−1. The remaining 34 patients received copanlisib 0.8 mg kg−1 plus CisGem, 20 of whom comprised the BTC expansion cohort.
Dose escalation and safety
The mean copanlisib dose was 46.0 mg in patients receiving copanlisib 0.6 mg kg−1 and the mean copanlisib dose ranged from 52.5 to 57.4 mg for the three cohorts receiving copanlisib 0.8 mg kg−1. Median duration of copanlisib treatment overall was 6.1 weeks (range 0.1–90.1) or two cycles (range 1–30). One patient in the BTC expansion cohort achieved 30 cycles.
Of the eight patients in the copanlisib 0.6 mg kg−1 plus gemcitabine dose-escalation cohort, six were evaluable for DLT assessment (one withdrawing because of clinical progression and one because of non-drug-related grade 2 fatigue and thrombocytopenia in cycle 1). One DLT was reported (grade 3 posterior reversible encephalopathy syndrome attributed to copanlisib and gemcitabine) and copanlisib dose escalation continued to 0.8 mg kg−1.
Of the eight patients in the copanlisib 0.8 mg kg−1 plus gemcitabine dose-escalation cohort, two were withdrawn during cycle 1 because of grade 2 abdominal pain assessed as copanlisib-related and grade 3 fatigue assessed as related to both copanlisib and gemcitabine, the latter fulfilling the DLT criteria. Copanlisib 0.8 mg kg−1+gemcitabine 1000 mg m−2 weekly in a 3 weeks on/1 week off schedule was the MTD and RP2D of this combination.
Fourteen patients were enrolled in the initial copanlisib 0.8 mg kg−1 plus CisGem cohort. One patient did not receive any copanlisib because of serious AEs (grade 3 abdominal pain and grade 3 non-cardiac chest pain) following the first administration of CisGem, and treatment was permanently discontinued. Two patients withdrew because of grade ⩾3 AEs assessed as related to copanlisib (grade 3 hypertension and grade 3 fatigue). In the subsequent expansion cohort of 20 patients with BTC, three withdrew because of grade ⩾3 AEs assessed as related at least to copanlisib (grade 4 sinus tachycardia, grade 3 non-cardiac chest pain and grade 4 decreased neutrophil count). Based on the evaluation of both cohorts, the RP2D of copanlisib plus CisGem was copanlisib 0.8 mg kg−1+cisplatin 25 mg m−2+gemcitabine 1000 mg m−2 weekly in a 2 weeks on/1 week off schedule.
All patients experienced at least one treatment-emergent AE (TEAE) (Supplementary Table S1), the most common being nausea (86%), hyperglycaemia (80%), decreased platelet count (80%), decreased neutrophil count (78%) and fatigue (76%).
Overall, 48 patients (96%) experienced at least one grade ⩾3 TEAE, most commonly decreased neutrophil count (62%), hypertension (42%), decreased platelet count (38%), hyperglycaemia (24%) and fatigue (22%). Two patients experienced a grade 5 TEAE (death): respiratory failure attributed to disease progression and heart failure not attributed to disease progression in a patient with a history of myocardial infarctions. Three additional patients died more than 30 days after discontinuation of study treatment, with the primary cause being AEs associated with clinical disease progression in two patients and disease progression in one patient. No deaths were considered related to study treatment.
Forty-seven patients (94%) experienced at least one copanlisib-related TEAE, most commonly hyperglycaemia (76%), nausea (74%), fatigue (66%), decreased neutrophil count (62%), decreased platelet count (58%), anorexia (56%), hypertension (46%), anaemia (42%) and vomiting (36%). Of grade ⩾3 copanlisib-related AEs, decreased neutrophil count (48%), hypertension (38%) and decreased platelet count (28%) were the most frequently observed (Table 2). Infusion-related increases in plasma glucose and plasma insulin (any grade) were observed in nearly all patients, and 14 patients received insulin as remedial drug therapy. The majority of infusion-related increases in plasma glucose and plasma insulin were self-limited and resolved within 24–48 h of copanlisib administration. Ten patients (20%) experienced a TEAE of grade 3 hyperglycaemia and two (4%) experienced grade 4 (Table 2).
Overall, 26 patients (52%) experienced a serious AE, most commonly lung infection (six patients, 12%), abdominal pain (four patients, 8%) and dehydration (three patients, 6%). Grade 4 serious TEAEs included sepsis (two patients, 4%) and lung infection, hyponatremia, anaemia, decreased neutrophil count, decreased platelet count, thromboembolic event, atrial fibrillation and sinus tachycardia (one patient each). Eleven patients (22%) experienced at least one serious copanlisib-related AE, including one patient (2%) with grade 4 sinus tachycardia and grade 4 sepsis.
Dose modifications (interruption or dose reduction) for copanlisib, gemcitabine or cisplatin due to TEAEs were necessary in 45 patients (90%); 26 (52%) had a dose modification because of copanlisib-related TEAEs. Overall, nine patients (18%) permanently discontinued copanlisib because of TEAEs, with fatigue (three patients) being the most common.
Pharmacokinetic evaluation
Forty-seven patients (94%) were valid for PK analysis.
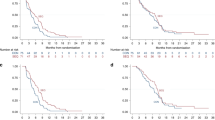
Following infusion of copanlisib on cycle 1, day 1 at both doses in combination with gemcitabine or copanlisib 0.8 mg kg−1 in combination with CisGem, geometric mean Cmax of copanlisib in plasma was between 249 and 414 μg l−1 at a median time to reach tmax of about 1 h (Figure 1A). Copanlisib demonstrated dose-proportional increase in plasma exposure (Cmax and AUC) over the dose range of 0.6–0.8 mg kg−1 (Figure 2). Geometric mean terminal half-life for copanlisib was between 25 and 26 h. Interpatient variability of copanlisib exposure (coefficient of variation) ranged from 34 to 88% (Supplementary Table S2).

Plasma concentrations vs time profiles of copanlisib and its metabolite M-1. Geometric mean/s.d. concentrations vs time profiles of copanlisib (μg l−1) (A) and its metabolite M-1 (μg l−1) (B) in plasma after copanlisib dosing on cycle 1, day 1 (the lower figures of each panel show the first 25 h after the start of the infusion). BTC=biliary tract cancer; CisGem=cisplatin (25 mg m−2) plus gemcitabine (1000 mg m−2).

Dose-normalised plasma pharmacokinetic parameters of copanlisib. Boxplots of dose-normalised parameters Cmax/dose (A), AUC(0–25)/dose (B) and AUC(0–tlast)/dose (C) of copanlisib in plasma following administration of copanlisib on cycle 1, day 1. AUC=area under the plasma concentration vs time curve; AUC(0–25)=AUC from 0 to 25 h after the start of the infusion; AUC(0–tlast)=area under the plasma concentration vs time curve from 0 to time of last observed plasma concentration value above the lower limit of quantification; BTC=biliary tract cancer; Cmax=maximum observed plasma concentration after single-dose administration; CisGem=cisplatin (25 mg m−2) plus gemcitabine (1000 mg m−2).
No substantial differences in plasma exposure (Cmax and AUC(0–25)) of copanlisib were observed upon co-administration of CisGem, and PK parameters did not differ considerably over the different dosing regimens (Supplementary Table S2).
Copanlisib metabolite M-1 was measured only at the 0.8 mg kg−1 dose. Geometric mean Cmax for M-1 ranged between 5.2 and 9.3 μg l−1 at a median tmax of 2–4 h (Figure 1B and Supplementary Table S3). Estimated geometric mean terminal half-life was comparable with that of copanlisib. Interpatient variability of metabolite M-1 exposure was high, with coefficient of variation ranging from 66 to 115% (Supplementary Table S3). The metabolite ratio of copanlisib metabolite M-1 to copanlisib in plasma for AUC(0–25) ranged from 1.5 to 19%. The amount of copanlisib and metabolite M-1 excreted into urine during the 25 h urine collection interval was low, with about 4.9% and 0.9%, respectively, of dose excreted.
The PK of total and free platinum, gemcitabine and its metabolite 2’,2’-difluorodeoxyuridine were not influenced by co-administration with copanlisib (further details provided in the Supplementary Methods section).
Copanlisib is a low renally excreted drug and a strong inhibitor of multidrug and toxin extrusion protein 2K (MATE2-K) in vitro. Patients treated with copanlisib plus cisplatin, a known substrate of MATE2-K, showed no signs of increased nephrotoxicity caused by intrarenal accumulation of cisplatin, suggesting a low potential effect of copanlisib on MATE2-K (data not shown). In addition, the plasma exposure of cisplatin measured in combination with copanlisib was in the range of previous historical PK observed for cisplatin (Himmelstein et al, 1981).
Efficacy analysis
All 50 patients were evaluable for efficacy and 40 (80%) underwent imaging for post-baseline RECIST assessment and were evaluable for best overall tumour response. One patient (2%) achieved a complete response, four (8%) achieved a partial response and 19 (38%) had stable disease, resulting in an objective response rate of 10% overall.
Of the 16 patients who received copanlisib plus gemcitabine, the response rate was 6.3% (1 out of 16 patients). One patient achieved a partial response (copanlisib dose of 0.6 mg kg−1; primary peritoneal carcinoma), discontinuing the study because of disease progression after eight cycles.
The response rate in patients who received copanlisib plus CisGem was 12% (4 out of 34 patients). One patient achieved a complete response (gallbladder cancer) and three patients achieved a partial response (intrahepatic biliary cancer) (Figure 3). Two patients who achieved a partial response were in the BTC expansion cohort, giving a response rate of 10% in that cohort (2 out of 20 patients).

Waterfall plot of best change in target lesion size from baseline vs BRAF, KRAS and PIK3CA mutation status and PTEN protein status for all patients with data for both biomarkers and change in target lesion size. ACC=adenoid cystic cancer; BC=breast cancer; BTC=biliary tract cancer; CC=cholangiocarcinoma; CisGem=cisplatin (25 mg m−2) plus gemcitabine (1000 mg m−2); Eso=oesophageal cancer; GIST=gastrointestinal stromal tumour; GC=gallbladder cancer; gem=gemcitabine (1000 mg m−2); IHC=immunohistochemistry; LS=liposarcoma; NGS=next-generation sequencing; NSCLC = non-small-cell lung cancer; Pa=pancreatic cancer; Pe=peritoneal cancer; SGC=salivary gland cancer (parotid gland). *The majority of patients with disease progression progressed because of non-target lesion growth.
Of the 23 patients with BTC overall, the response rate was 17% (4 out of 23 patients). All four responders had not received any prior anti-cancer therapy, giving a response rate of 36% in the 11 patients with BTC who had not received prior anti-cancer therapy. No response was observed in the 12 patients who had received prior CisGem or gemcitabine-containing chemotherapy.
Further efficacy details are provided in the Supplementary Methods section.
Biomarkers
Mutation data from circulating tumour DNA were generated from 43 out of 50 patients, and 22 out of 43 patients had tumour next-generation sequencing data. PIK3CA alterations were detected in 2% of patients (1 out of 43; a gene amplification), KRAS mutations in 23% (10 out of 43) and BRAF mutations in 9% (2 out of 22). Complete tumour PTEN protein loss was identified in 41% of patients (12 out of 29). Among patients with BTC, the mutation rates for PIK3CA, KRAS and BRAF were 0% (0 out of 22), 18% (4 out of 22) and 22% (2 out of 9), respectively, and PTEN protein loss was observed in 69% of patients (9 out of 13). No mutations in PTEN were identified by next-generation sequencing.
Two patients with objective tumour response had PTEN protein loss, one in conjunction with a KRAS mutation (patient with cholangiocarcinoma; BTC expansion cohort) and the other with a BRAF mutation (patient with cholangiocarcinoma; initial CisGem safety evaluation cohort) (Figure 3). No evidence of molecular aberration in these factors was observed in the other three patients with tumour response. No biomarker was clearly associated with outcome, although not all biomarkers could be assayed in all patients because of limitations of sample availability and appropriate informed consent.
Discussion
This phase I, open-label study determined the MTD and RP2D of copanlisib plus gemcitabine and the RP2D of copanlisib plus CisGem. The MTD and RP2D of copanlisib plus gemcitabine 1000 mg m−2 was 0.8 mg kg−1 on days 1, 8 and 15 of a 28-day treatment cycle. The one DLT reported was posterior reversible encephalopathy syndrome, attributed to copanlisib and gemcitabine, which has previously been associated with gemcitabine (Rajasekhar and George, 2007; Marrone et al, 2011). The RP2D for the triplet combination was copanlisib 0.8 mg kg−1+gemcitabine 1000 mg m−2+cisplatin 25 mg m−2 on days 1 and 8 of a 21-day treatment cycle.
Copanlisib plus CisGem demonstrated an acceptable safety profile. Most toxicities were manageable through dose modifications and remedial drug treatment. No new or unexpected safety signals were observed. The most common TEAEs were nausea, hyperglycaemia and decreased platelet count. Most patients experienced at least one grade ⩾3 TEAE, most commonly decreased neutrophil count, hypertension and decreased platelet count. The observed safety profile of copanlisib plus CisGem was generally consistent with previously observed toxicities of copanlisib monotherapy (hyperglycaemia, nausea and hypertension), although the incidence of haematological TEAEs was higher than previously reported with either gemcitabine or copanlisib alone (Valle et al, 2010; Patnaik et al, 2016). This is likely to be related to the combination of the well-known safety profiles of gemcitabine (haematological toxicities and flu-like symptoms) and cisplatin (haematological toxicities and nausea) (Valle et al, 2010; Lilly USA, LLC, 2014).
The acceptable safety profile of the copanlisib plus CisGem combination is in contrast to that of the approved PI3K-δ inhibitor idelalisib, which demonstrated an increased rate of AEs, including deaths, in combination with other anti-cancer therapies in clinical trials, resulting in the termination of six trials in patients with chronic lymphocytic leukaemia, small lymphocytic lymphoma and indolent non-Hodgkin’s lymphomas (US Food and Drug Administration, 2016).
Infusion-related increases in plasma glucose and plasma insulin were observed in the majority of patients (∼80%), in line with the expected mechanism of action and consistent with observations from previous clinical studies (Patnaik et al, 2016; Dreyling et al, 2017a). Few patients received systemic corticosteroids as concomitant medication; therefore, the observed increase in glucose is likely attributed to copanlisib. Although a direct analysis of the correlation between copanlisib exposure or tumour response and plasma glucose or insulin levels was not performed and some confounding factors such as corticosteroid and insulin administrations were present, plasma glucose and plasma insulin remain useful pharmacodynamic markers of on-target copanlisib exposure.
Copanlisib exhibited favourable PK properties in combination with CisGem, with rapid absorption, a nearly dose-proportional increase in exposure in the dose-escalation cohorts and minimal differences between the two drug combinations tested. There was no indication of any clinically relevant PK interactions between copanlisib and gemcitabine or cisplatin, consistent with the known metabolic pathway for copanlisib.
Recent analyses indicate that an intermittent dose schedule of 60 mg weekly on days 1, 8 and 15 of a 28-day cycle was likely to achieve a similar risk–benefit ratio as 0.8 mg kg−1 weight-based dosing (Reif et al, 2016), and fixed dosing is being explored in ongoing studies.
The objective response rate was 10% overall, 12% with the copanlisib plus CisGem combination and 6.3% with the copanlisib plus gemcitabine combination; however, many of these patients had already been treated with CisGem as first-line therapy. The response rate was 36% in patients with BTC with no prior anti-cancer therapy (4 out of 11 patients). BTC is a heterogeneous group of malignancies, so direct comparison with other studies is subject to selection bias. A large phase III study of patients with advanced BTC receiving CisGem demonstrated a response rate of 26% (Valle et al, 2010). Further investigation of copanlisib plus CisGem is warranted.
The exploratory biomarker analysis observed PTEN loss in 41% of evaluable patients (69% in patients with BTC). This observation is in line with previous reports in gallbladder cancer, where PTEN loss was found in 72% of patients (Ali et al, 2015), suggesting that this tumour type is widely reliant on PI3K pathway signalling through this mechanism. The prevalence of KRAS mutations detected in patients with BTC (18%) is also similar to what has been previously reported (∼20%), whereas the PIK3CA mutation rate (0%) is lower and the BRAF mutation rate (22%) is higher than what has been reported in previous studies (∼10% and 5%, respectively) (Jain and Javle, 2016; Javle et al, 2016; Zhao et al, 2016). Although none of the biomarkers evaluated associated clearly with outcome in this cohort, these analyses may be limited by the mixed tumour types and small numbers of patients included in the molecular subgroups.
In summary, copanlisib demonstrated an acceptable safety profile and potentially promising clinical response when administered at a dose of 0.8 mg kg−1 (approximately equivalent to 60 mg fixed dose) in combination with CisGem. Pharmacokinetic profiles for copanlisib and its metabolite M-1 were favourable, and co-administration of CisGem had no influence on copanlisib and metabolite M-1 exposure. A phase II study investigating the clinical benefits of copanlisib plus CisGem in patients with advanced cholangiocarcinoma (NCT02631590) is currently underway.
References
Ali A, Mishra PK, Sharma S, Arora A, Saluja SS (2015) Effects of PTEN gene alteration in patients with gallbladder cancer. Cancer Genet 208: 587–594.
Deshpande V, Nduaguba A, Zimmerman SM, Kehoe SM, MacConaill LE, Lauwers GY, Ferrone C, Bardeesy N, Zhu AX, Hezel AF (2011) Mutational profiling reveals PIK3CA mutations in gallbladder carcinoma. BMC Cancer 11: 60.
Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D (2006) BEAMing: single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods 3: 551–559.
Dreyling M, Morschhauser F, Bouabdallah K, Bron D, Cunningham D, Assouline SE, Verhoef G, Linton K, Thieblemont C, Vitolo U, Hiemeyer F, Giurescu M, Garcia-Vargas J, Gorbatchevsky I, Liu L, Koechert K, Peña C, Neves M, Childs BH, Zinzani PL (2017a) Phase II study of copanlisib, a PI3K inhibitor, in relapsed or refractory, indolent or aggressive lymphoma. Ann Oncol 28: 2169–2178.
Dreyling M, Santoro A, Mollica L, Leppa S, Follows GA, Lenz G, Kim WS, Nagler A, Panayiotidis P, Demeter J, Ozcan M, Kosinova M, Bouabdallah K, Morschhauser F, Stevens DA, Trevarthen D, Giurescu M, Cupit L, Liu L, Kochert K, Seidel H, Pena C, Yin S, Hiemeyer F, Garcia-Vargas J, Childs BH, Zinzani PL (2017b) Phosphatidylinositol 3-kinase inhibition by copanlisib in relapsed or refractory indolent lymphoma. J Clin Oncol 35: 3898–3905.
Ebata T, Ercolani G, Alvaro D, Ribero D, Di Tommaso L, Valle JW (2017) Current status on cholangiocarcinoma and gallbladder cancer. Liver Cancer 6: 59–65.
Hägebarth A, Haike K, Paul J, Mumberg D, Ziegelbauer K, Liu N (2012) Potent in vitro and in vivo antitumor activity of PI3K inhibitor BAY 80-6946 and MEK inhibitor BAY 86-9766 in preclinical biliary tract cancer models. Annual Meeting of the American Association for Cancer Research, Chicago, Illinois, USA, 31 March–4 April, Poster 869.
Haike K, Stasik E, Soujon M, Wengner AM, Petrova E, Genvresse I, Wilhelm S, Childs BH, Mumberg D, Liu N (2014) Molecular mechanisms supporting inhibition of PI3K isoforms by copanlisib in blocking B-cell signaling and tumor cell growth in diffuse large B-cell lymphoma. 1st American Society of Hematology Meeting on Lymphoma Biology, Colorado Springs, Colorado, USA, 10–13 August, Poster 48.
Himmelstein KJ, Patton TF, Belt RJ, Taylor S, Repta AJ, Sternson LA (1981) Clinical kinetics on intact cisplatin and some related species. Clin Pharmacol Ther 29: 658–664.
Ihle NT, Powis G (2009) Take your PIK: phosphatidylinositol 3-kinase inhibitors race through the clinic and toward cancer therapy. Mol Cancer Ther 8: 1–9.
Jain A, Javle M (2016) Molecular profiling of biliary tract cancer: a target rich disease. J Gastrointest Oncol 7: 797–803.
Javle M, Bekaii-Saab T, Jain A, Wang Y, Kelley RK, Wang K, Kang HC, Catenacci D, Ali S, Krishnan S, Ahn D, Bocobo AG, Zuo M, Kaseb A, Miller V, Stephens PJ, Meric-Bernstam F, Shroff R, Ross J (2016) Biliary cancer: utility of next-generation sequencing for clinical management. Cancer 122: 3838–3847.
Lee JJ, Loh K, Yap YS (2015) PI3K/Akt/mTOR inhibitors in breast cancer. Cancer Biol Med 12: 342–354.
Li X, Wu C, Chen N, Gu H, Yen A, Cao L, Wang E, Wang L (2016) PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 7: 33440–33450.
Lilly USA, LLC (2014) Gemzar (gemcitabine) (package insert). Available at: http://pi.lilly.com/us/gemzar.pdf . Accessed 16 June 2017.
Liu N, Rowley BR, Bull CO, Schneider C, Haegebarth A, Schatz CA, Fracasso PR, Wilkie DP, Hentemann M, Wilhelm SM, Scott WJ, Mumberg D, Ziegelbauer K (2013) BAY 80-6946 is a highly selective intravenous PI3K inhibitor with potent p110α and p110δ activities in tumor cell lines and xenograft models. Mol Cancer Ther 12: 2319–2330.
Marrone LC, Marrone BF, de la Puerta Raya J, Gadonski G, da Costa JC (2011) Gemcitabine monotherapy associated with posterior reversible encephalopathy syndrome. Case Rep Oncol 4: 82–87.
Patnaik A, Appleman LJ, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Weiss GJ, Sachdev JC, Chadha M, Fulk M, Ejadi S, Mountz JM, Lotze MT, Toledo FGS, Chu E, Jeffers M, Peña C, Xia C, Reif S, Genvresse I, Ramanathan RK (2016) First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin's lymphomas. Ann Oncol 27: 1928–1940.
Rajasekhar A, George TJ Jr (2007) Gemcitabine-induced reversible posterior leukoencephalopathy syndrome: a case report and review of the literature. Oncologist 12: 1332–1335.
Reif S, Ahsman M, Jentsch G, Wiegert E, Grevel J, Granvil C (2016) Use of a population pharmacokinetic approach and time-to-event analysis to support the clinical recommendation of a flat dosing of copanlisib in cancer patients. Clin Pharmacol Ther 99: pS5.
Singh SS, Yap WN, Arfuso F, Kar S, Wang C, Cai W, Dharmarajan AM, Sethi G, Kumar AP (2015) Targeting the PI3K/Akt signaling pathway in gastric carcinoma: a reality for personalized medicine? World J Gastroenterol 21: 12261–12273.
US Food and Drug Administration (2016) FDA Alerts Healthcare Professionals About Clinical Trials with Zydelig (idelalisib) in Combination with other Cancer Medicines. Available at: https://www.fda.gov/Drugs/DrugSafety/ucm490618.htm . Accessed 16 June 2017.
Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, Madhusudan S, Iveson T, Hughes S, Pereira SP, Roughton M, Bridgewater J for the ABC-02 Trial Investigators (2010) Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 362: 1273–1281.
Wang X, Huang H, Young KH (2015) The PTEN tumor suppressor gene and its role in lymphoma pathogenesis. Aging 7: 1032–1049.
Yuan TL, Cantley LC (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27: 5497–5510.
Zhao S, Cao Y, Liu SB, Wang XA, Bao RF, Shu YJ, Hu YP, Zhang YJ, Jiang L, Zhang F, Liang HB, Li HF, Ma Q, Xu Y, Wang Z, Zhang YC, Chen L, Zhou J, Liu YB (2016) The E545K mutation of PIK3CA promotes gallbladder carcinoma progression through enhanced binding to EGFR. J Exp Clin Cancer Res 35: 97.
Acknowledgements
Medical writing assistance was provided by Laura Badtke, PhD (Complete HealthVizion, Chicago, IL, USA), based on detailed discussion and feedback from all the authors. We thank Ron Dubowy, Camille Granvil and Jie Cheng.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
RDK reports grants and personal fees from Bayer and Bristol-Myers Squibb, personal fees from Lilly and grants from Eisai. JEG-O reports institutional clinical trial support from Bayer, Novartis, Peregrine Pharmaceuticals, NanoCarrier, Genentech, Seattle Genetics, MedImmune and Pfizer. CP, CX and AK are employees of Bayer HealthCare. IG and AA-H are employees of Bayer AG. SRA declares no competing financial interest.
Additional information
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Kim, R., Alberts, S., Peña, C. et al. Phase I dose-escalation study of copanlisib in combination with gemcitabine or cisplatin plus gemcitabine in patients with advanced cancer. Br J Cancer 118, 462–470 (2018). https://doi.org/10.1038/bjc.2017.428
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.428
Keywords
This article is cited by
-
PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer
Molecular Cancer (2023)
-
Phase Ib Trial of the PI3K Inhibitor Copanlisib Combined with the Allosteric MEK Inhibitor Refametinib in Patients with Advanced Cancer
Targeted Oncology (2020)
-
Targeting PI3K in cancer: mechanisms and advances in clinical trials
Molecular Cancer (2019)
-
Weighing the prognostic role of hyponatremia in hospitalized patients with metastatic solid tumors: the HYPNOSIS study
Scientific Reports (2019)
-
The PI3K inhibitor copanlisib synergizes with sorafenib to induce cell death in hepatocellular carcinoma
Cell Death Discovery (2019)
