
Abstract
Background:
Elevated body mass index (BMI, kg m−2) has been consistently associated with oesophageal adenocarcinoma (EA) and gastric cardia adenocarcinoma (GCA) incidence. However, effects of adiposity over the life course in relation to EA/GCA have not been thoroughly explored.
Methods:
We pooled two prospective cohort studies: NIH-AARP Diet and Health Study and Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial, with data on 409 796 individuals (633 EA, 415 GCA). At baseline, participants reported their height and weight at ages 20 and 50 years, and current. Body mass index trajectories were determined using latent class analysis. Hazard ratios (HRs) and 95% confidence intervals (CI) were estimated using proportional hazards regression.
Results:
Compared with individuals with a BMI<25 kg m−2 at all time points, exceeding a BMI of 25 kg m−2 at age 20 was associated with increased risks of EA (HR=1.76, 95% CI: 1.35–2.29) and GCA (HR=1.62, 95% CI: 1.16–2.25). Similarly, a BMI trajectory of overweight (⩾25–<30 kg m−2) at age 20 progressing to obesity (⩾30 kg m−2) by age 50 was associated with increased risks of EA (HR=2.90, 95% CI: 1.67–5.04) and GCA (HR=4.07, 95% CI: 2.32–7.15), compared with individuals with a normal weight (⩾18.5–<25 kg m−2) trajectory. Weight gain of ⩾20 kg between age 20 and baseline was also associated with a two times increased risk of EA (HR=1.97, 95% CI: 1.43–2.73) and more modestly with GCA (HR=1.40, 95% CI: 0.96–2.05).
Conclusions:
Being overweight in early adulthood and weight gain later in life were each associated with increased risks of EA and GCA. This underscores the potential of weight control programs for reducing EA and GCA risk.
Similar content being viewed by others


Main
Over the last two decades, oesophageal and gastric cardia adenocarcinomas (EA/GCA) have been among the most rapidly increasing cancer types in the USA and in other Western countries (Devesa et al, 1998; Simard et al, 2012). Oesophageal adenocarcinoma and GCA have overlapping pathogeneses, they are both glandular epithelial cancers originating in or near the gastroesophageal junction, and they have similar risk factor profiles and 5-year survival rates of ∼26% (Wijnhoven et al, 1999). Increasing rates of obesity in the general population have generally paralleled the increasing rates of EA/GCA. Elevated body mass index (BMI, kg m−2) has consistently been shown to be associated with incidence of EA and GCA (Hoyo et al, 2012). Although the underlying causal mechanisms that result in altered cancer risks are unclear, leading hypotheses include the ideas that high levels of adiposity may promote gastroesophageal reflux disease (Wild and Hardie, 2003) and/or metabolic sequelae (Reid et al, 2003; Ryan et al, 2011).
Meta-analyses have reported associations between BMI, waist circumference, and waist-to-hip ratio and increased EA/GCA risk (Hoyo et al, 2012; Singh et al, 2013). However, few studies to date have examined early adulthood body weight (Chow et al, 1998; Lagergren et al, 1999; Cheng et al, 2000; Wu et al, 2001; Merry et al, 2007) and body weight change (Chow et al, 1998; Merry et al, 2007); all reported increased risks of EA/GCA with early adulthood obesity and weight gain. One study examined the association between trajectories of body shape, but not BMI, and EA risk. There was a suggestive association between the trajectories where individuals reported being lean in early life and heavy in later life or remaining heavy throughout their life and increased EA risk. However, this study had a small EA sample size (n=98) and did not examine GCA (Song et al, 2015). As the prevalence of obesity (BMI⩾30 kg m−2) is expected to continue to increase (Finkelstein et al, 2012), it is vital to understand the timing of obesity and weight gains in relation to EA and GCA outcomes. This may provide mechanistic insight and inform the time window of when interventions may be successfully implemented to reduce the incidence of these highly lethal cancers.
In the current study, we pooled data from the National Institutes of Health (NIH)-AARP Diet and Health Study with the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial to examine the relationship between early adulthood BMI, weight change, average BMI, and BMI trajectories and risk of EA/GCA.
Materials and methods
Study population
We utilised data from two study populations – the NIH-AARP Diet and Health Study, and the PLCO Cancer Screening Trial. These studies were combined because the inclusion criteria and available data were very similar. Thus, data variables could be harmonised, allowing for a larger sample size to examine the rare outcomes of EA and GCA. Both study protocols were approved by the Institutional Review Board of the National Cancer Institute.
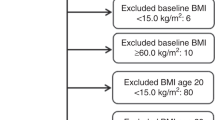
The NIH-AARP Diet and Health Study is a prospective cohort study conducted in six states (California, Florida, Louisiana, New Jersey, North Carolina, and Pennsylvania) and two metropolitan areas (Atlanta, Georgia and Detroit, Michigan; Schatzkin et al, 2001). Eligible participants were male and female AARP members, aged 50–71 years at the time of study recruitment. In 1995–1996, eligible members were mailed a baseline questionnaire, which assessed current anthropometric measurements, demographics, diet, and medical history. Six months later, participants who completed the baseline questionnaire and did not self-report colon, breast, or prostate cancer were mailed a risk factor questionnaire (RFQ), which asked about additional exposures including recalled anthropometric measurements. Participants who completed the RFQ were more likely to be female, white, and have a college degree, compared to individuals whom completed only the baseline questionnaire (Supplementary Table S1). Of the 337 071 participants who completed the RFQ, individuals who died (n=1619) or moved (n=547) before the RFQ was scanned were excluded. In addition, individuals with a proxy report on baseline or RFQ (n=10 383), cancer diagnosis prior to RFQ (n=18 721), zero follow-up time (n=30), or death report only of cancer (n=2451) were excluded from our analysis. Finally, we excluded individuals with a missing value for BMI at age 18, 50, or baseline (n=28 686) or with implausible BMI values (<15 or >60 kg m−2, n=3363). Our final analytic cohort from the NIH-AARP Study consisted of 271 271 individuals.
The PLCO Cancer Screening Trial is a randomised controlled trial conducted at 10 study sites (Washington, DC; Detroit, Michigan; Marshfield, Wisconsin; Honolulu, Hawaii; Birmingham, Alabama; Aurora, Colorado; Minneapolis, Minnesota; Pittsburgh, Pennsylvania; Boise, Idaho; and St Louis, Missouri; Kramer et al, 1993). Eligible participants were men and women, aged 50–79 years at the time of study recruitment. In 1993–2001, eligible participants completed a self-administered baseline questionnaire, which assessed anthropometric measurements, demographics, and medical history. Of the 149 943 participants who completed the baseline questionnaire, individuals with a prior cancer diagnosis (n=6907), self or death report of cancer only (n=192), zero follow-up time (n=795), or age <50 years (n=1) were excluded from our analysis. In addition, we excluded individuals with a missing value for BMI at age 20, 50, or baseline (n=3462) or with implausible BMI values (<15 or >60 kg m−2, n=61). Our final analytic cohort from the PLCO Cancer Screening Trial consisted of 138 525 individuals.
Outcomes
Study participants were followed for incident primary diagnoses of histologically verified EA or GCA through 31 December 2011, for NIH-AARP (Schatzkin et al, 2001) and 31 December 2009, for PLCO (Kramer et al, 1993). Oesophageal cancer (defined as International Classification of Disease for Oncology, 3rd edition [ICD-O-3] diagnostic code C15.0–C15.9) and gastric cardia cancer (ICD-O-3 diagnostic code C16.0) for NIH-AARP participants was ascertained by linkage to state cancer registries (including states of participant recruitment and Arizona, Nevada, and Texas), which is estimated to capture ∼90% of all cancer cases (Michaud et al, 2005), and for PLCO participants was ascertained through self-report and subsequent medical records verification. Cases were then classified as adenocarcinoma (ICD-O-3 histology codes of 8140-8575). The current analysis included 633 EA cases, 415 GCA cases, and 408 748 non-cases.
Exposures
Both studies asked participants to report their current height and weight at baseline. Participants also reported recalled weight at ages 18 (NIH-AARP) or 20 (PLCO) years – for simplicity we will refer to this as age 20 for both studies – and 50 years. The NIH-AARP questionnaire additionally asked participants to report maximal weight. Body mass index, calculated as weight in kilograms (kg) divided by height in metres (m) squared (kg m−2), was examined as a continuous metric and as a categorical metric, the latter according to World Health Organisation definitions: underweight (<18.5), normal weight (18.5–24.9), overweight (25.0–29.9), and obese (⩾30.0; WHO, 2000).
Body mass index was assessed at discrete age points – age 20 years, age 50 years, and age at baseline. We also examined age when BMI first exceeded 25 kg m−2 (Adams et al, 2014). In addition, we examined both average BMI, between these age points, and maximal BMI as predictors. Weight change between these ages was defined as ⩽−2, >−2 to <5 (stable weight, referent), 5 to <15, and ⩾15 kg (de Mutsert et al, 2014). To further explore excess weight gain, we examined an additional model where the highest category of weight change was defined as ⩾20 kg. Thus, the categories were defined as ⩽2, >2–<5, 5–<15, 15–<20, and ⩾20 kg.
Body mass index trajectories were determined utilising latent class group-based mixture model analysis (PROC TRAJ, SAS 9.3, SAS Institute, Cary, NC, USA) (Jones and Nagin, 2007). This allowed us to group individuals with similar BMI patterns between the ages of 20, 50, and baseline. We allowed 3–5 trajectory categories as well as linear or quadratic patterns of BMI change. The optimal number of categories and model pattern was selected using the change in Bayesian Information Criterion and the mean posterior probability of each trajectory, where each trajectory grouping was required to include at least 1% of participants. Thus, we selected four trajectory categories (stable normal BMI, normal BMI to overweight, normal BMI to obese, and overweight to obese), with quadratic patterns of BMI change.
Statistical analysis
Cox proportional hazard regression analysis was used to calculate adjusted hazard ratios (HRs) and 95% confidence intervals (CIs) for the association of adiposity with EA and GCA, with age as the underlying time metric. Follow-up of the analytic cohort occurred from time at completion of RFQ questionnaire (NIH-AARP) or baseline questionnaire (PLCO) until an event (i.e., incident EA or GCA) or right censoring (i.e., other cancer diagnosis, death, loss to follow-up, or last date of follow-up), whichever occurred first. In models examining GCA as the primary outcome, EA was a censored event, and vice versa. The proportional hazards assumption was tested by examining interactions between BMI (defined as continuous and categorical) and log (time) in models that included confounders; no violation of this assumption was observed (P⩾0.05).
Effect modification by sex, cigarette smoking, diabetes, physical activity, and study was assessed using likelihood ratio tests that compared regression models with and without a multiplicative term (Kleinbaum, 2002). Potential confounders (Rothman et al, 2008) included alcohol consumption, aspirin and NSAID use, diabetes, education, fruit/vegetable intake, marital status, physical activity, race/ethnicity, red/white meat intake, smoking, and total energy intake. Variables remained in the adjusted model if they were associated with the exposure and outcome, and variable elimination changed the log HR by ⩾10% (Rothman et al, 2008); sex, race (non-Hispanic White, non-Hispanic Black, other), smoking (never/current/former, and categorised cigarettes per day (0, 1–10, 11–20, 21–30, 31–40, 41–60, 61+)), and education (high school or less, technical school or some college, college graduate, postgraduate) met this criterion and were included in all final models. We adjusted for study in all models. Models for weight were additionally adjusted for height. Analyses were conducted using SAS version 9.3. Tests of linear trend were conducted using continuous variables. All P-values are two-sided.
Sensitivity analyses
As a sensitivity analysis, we adjusted weight change models for average weight (Oldham, 1962; Tu and Gilthorpe, 2007), to determine if weight gain increased the risk of EA/GCA beyond what is conferred by average lifetime weight alone.
Results
Demographic characteristics of EA and GCA cases, and non-cases are shown in Table 1. Compared with non-cases, individuals who developed EA or GCA were more likely to be older, male, non-Hispanic white, and have ever-smoked cigarettes.
Table 2 presents associations between BMI metrics – that is BMI at ages 20 and 50 years, age at baseline, maximal, and time at which BMI first exceeded the threshold of 25 kg m−2 – in relation to risks of EA and GCA. Overweight at age 20 was associated with increased risks of EA (HRoverweight=1.31, 95% CI: 1.06–1.63) and GCA (HRoverweight=1.39, 95% CI: 1.05–1.83), but obesity was not associated with a significantly increased risk of EA (HRobese=1.44, 95% CI: 0.85–2.46) or GCA (HRobese=1.02, 95% CI: 0.45–2.29). However, this could be due to small sample size in the obese category at age 20, as the trend is still significant. The associations between overweight and obesity at age 50 years and EA (HRoverweight=1.34, 95% CI: 1.11–1.61; HRobese=2.09, 95% CI: 1.66–2.65) and GCA (HRoverweight=1.39, 95% CI: 1.11–1.75; HRobese=1.63, 95% CI: 1.19–2.23) were similar to risk estimates for BMI at study baseline. The group of participants whom first exceed the threshold of 25 kg m−2 at age 20 had a 60–70% increased risk of EA and GCA (HR=1.76, 95% CI: 1.35–2.29; HR=1.62, 95% CI: 1.16–2.25, respectively), compared with individuals who had a BMI of <25 kg m−2 throughout their lifetime; associations were weaker among those first exceeding a BMI of 25 kg m−2 at age 50 or baseline age.
Early adulthood weight change (i.e., age 20–50 years) of 15 kg or more was associated with modest increased risks of EA (HR=1.26, 95% CI: 0.98–1.62). Risk estimates for EA were higher when a change of 15 kg occurred after age 50 (i.e., age 50 years to baseline; HR=1.57, 95% CI: 1.19–2.08). However, total lifetime weight change (i.e., age 20 years to baseline) was strongly associated with EA risk in a dose-response pattern culminating in a HR of 1.97 (95% CI: 1.43–2.73) for individuals who had gained 20 kg or more. Associations with GCA were generally similar to EA. When we further adjusted these models for average weight, the results were slightly attenuated, but estimates for the relationship between lifetime increases in weight and EA/GCA risk remained similar (Table 3).
Overweight or obese average BMI across all time points was associated with an increased risk of both cancer types (Table 4). Average lifetime BMI between age 20 and baseline of 30 kg m−2 or more was most strongly associated with EA risk (HR=2.51, 95% CI: 1.91–3.31), whereas a two times increased risk was seen at each time point for GCA risk.
In the BMI trajectory analysis, compared with individuals who remained at a normal BMI level throughout their lifetime  , individuals who progressed from overweight to obese
, individuals who progressed from overweight to obese  had a three times or more increased risk of EA and GCA (HR=2.90, 95% CI: 1.67–5.04; HR=4.07, 95% CI: 2.32–7.15, respectively). Increased risk of EA and GCA was also noted for groups of participants that increased from a normal BMI to overweight or obese (Figure 1 and Table 5).
had a three times or more increased risk of EA and GCA (HR=2.90, 95% CI: 1.67–5.04; HR=4.07, 95% CI: 2.32–7.15, respectively). Increased risk of EA and GCA was also noted for groups of participants that increased from a normal BMI to overweight or obese (Figure 1 and Table 5).

Latent class trajectories of body mass index (BMI), NIH-AARP Diet and Health Study, and PLCO Cancer Screening Trial.
We found little evidence of effect measure modification (P⩾0.05) by sex, cigarette smoking, diabetes, physical activity, or study. However, analyses stratified by sex and study are provided in Supplementary Tables S2–S17, respectively.
Discussion
In this pooled analysis of two large prospective studies, the results indicate that continued increases in excess weight across the life course, particularly when exposure begins by early adulthood, is associated with increased risks of EA and GCA. The strongest associations between adiposity and EA/GCA were observed for individuals who had a trajectory of overweight at the onset of adulthood and progressed to obesity in later life. Among this group of individuals, we found a three times or more increased risk of both EA and GCA. Similarly, individuals who first reported being overweight at age 20 had 60–70% increased risks of EA and GCA.
Our findings extend previous analyses, which have indicated that early adult weight or adult weight change are important in conferring increased risks for EA and GCA (Chow et al, 1998; Lagergren et al, 1999; Cheng et al, 2000; Wu et al, 2001; Merry et al, 2007; Cook et al, 2015) by using correct weight change model specification (Oldham, 1962; Tu and Gilthorpe, 2007) and trajectory modelling (Jones and Nagin, 2007). These approaches have enabled us to identify critical periods for obesity and weight gain, specifically early adult obesity and adult weight gain. One previous study, utilising data from the Nurses’ Health Study and Health Professionals Follow-Up Study, reported a two times increased risk of EA associated with a body shape trajectory of heavy-stable/increasing. However, this study had a small sample size to examine EA (n=98), and did not consider GCA (Song et al, 2015).
We report that a weight gain of ⩾20 kg between age 20 and baseline is associated with a 97% increased risk of EA, compared to stable weight. In contrast, a study utilising data from the Health Professionals Follow-up Study examined obesity-related cancers collectively (i.e., colorectal, renal, pancreatic, and oesophageal) and reported an 11% increase in risk of obesity-related cancer associated with ⩾15 kg gained between age 21 and baseline assessment (de Mutsert et al, 2014). Although the sample size in this prior study was limited and EA could not be examined as a distinct outcome, our findings of a stronger association between weight change from age 20 to baseline and EA could indicate that gaining weight during adulthood is more important for EA than other obesity-related cancer types. Our results are in line with two previous studies that have examined EA and GCA as distinct outcomes conducted in The Netherlands (Merry et al, 2007) and the USA (Chow et al, 1998). The Netherlands cohort study reported a 3.4 and 2.1 times increased risk of EA and GCA, respectively, associated with an 8 kg m−2 change in BMI between age 20 and cohort baseline (Merry et al, 2007). The US case–control study reported a 2.1 and 1.3 times higher risk of EA and GCA, respectively, for a ⩾21 kg weight gain between the 20s and typical adult weight (Chow et al, 1998).
For the sensitivity analysis, we present models of weight change adjusted for average weight, as these are orthogonal variables (Oldham, 1962). Simply adjusting a model of weight change for baseline weight can produce spurious results, as they are not independent variables (Oldham, 1962; Tu and Gilthorpe, 2007). For example, adjusting a model of weight change (e.g., between ages 20 and 50 years) for weight at age 50 is essentially equivalent to estimating the relationship of weight at age 20 with the outcome. The same problem would occur if the weight change model was adjusted for weight at age 20 – this model would essentially be estimating the relationship of weight at age 50 with the outcome. When we adjusted weight gain for average weight, we still observe an association between large weight gains and EA/GCA risk. These results indicate that lifetime weight gain increases the risk of EA and GCA beyond that conferred by average lifetime weight alone.
Although the exact mechanisms underlying the association between adiposity and EA/GCA are unclear, it is hypothesised that central adiposity may increase intra-abdominal pressure. This promotes gastroesophageal reflux disease, which is a risk factor for Barrett’s esophagus (Wild and Hardie, 2003), the precursor lesion of EA (Sharma et al, 2004). Alternatively, dysfunctional adipose tissue may lead to metabolic sequelae, such as glucose intolerance, hypertension, or metabolic syndrome, which may have carcinogenic effects resulting in increased risks of EA or GCA (Reid et al, 2010; Ryan et al, 2011).
In this study, overweight and obesity at all time points was associated with an increased risk of EA and GCA. However, the increased risks of these cancers associated with overweight or obesity at age 20 years suggests that childhood and adolescent adiposity may be an important factor for these cancer types. Although we don’t have data on childhood and adolescence adiposity, a recent study reported that childhood obesity is associated with an increased risk of EA (Cook et al, 2015), which may be mediated through the moderate correlations with adult obesity (Singh et al, 2008; Macfarlane et al, 2011). Thus, it is possible that some of the associations reported herein are due to ‘tracking’ (correlation) of adiposity from childhood through early and mid-adulthood. Alternatively, associations of childhood obesity with gastroesophageal reflux disease (Malaty et al, 2009; Pashankar et al, 2009; Ruigomez et al, 2010; Koebnick et al, 2011) and metabolic disorders may directly affect future risks of EA and GCA (Adegboye et al, 2010; Abrams and Levitt Katz, 2011).
This is the first study to examine BMI trajectories in relation to EA and GCA risks, but these trajectories are based on self-reported and recalled age-specific weights. However, recalled weight has been shown to be highly correlated with measured weight that was assessed 20 years prior (r=0.88; Dahl and Reynolds, 2013), and self-reported weight has been shown to be highly correlated with measured weight over repeated measurements (r=0.97; Dahl et al, 2010). Although these correlations are high, misclassification can occur across BMI categories (Spencer et al, 2002). Therefore, caution is needed when interpreting studies with self-reported body weight and height. In addition, we utilised BMI as a proxy for body fatness, which may be an inaccurate measurement of body fatness for an individual. However, percentage body fatness and BMI are strongly correlated within sex-age groups (r=0.72–0.84) (Flegal et al, 2009). Thus, BMI can be useful at the population level for distinguishing body fatness categories (Flegal et al, 2009). We also only had data at three time points – ages 20 and 50 years, and age at baseline. Future studies should prospectively collect height and weight at regular intervals for a thorough examination of the association between adiposity over the life course and cancer outcomes. In addition, these studies did not include information on some important risk factors for EA and GCA. Specifically, gastroesophageal reflux disease was not assessed, which may be an important stratification – some individuals’ risks of EA/GCA may be predominately mediated through gastroesophageal reflux as opposed to putative carcinogenic effects of metabolic disorders. Also, our study population predominately included participants of European ancestry. However, this is the group at highest risk of developing EA/GCA (Falk, 2009). Finally, the number of GCA cases may be underestimated, as a large proportion of gastric cancers with an overlapping or unknown location (i.e., C16.8-9) may be tumours of the cardia (Corley and Kubo, 2004).
This study utilised a pooled analysis to evaluate the association between adiposity and EA/GCA, which allowed for a larger sample size than an individual study alone. This allowed for examination of EA and GCA as distinct outcomes. However, even with a large sample size, we were limited in the number of BMI trajectories we could examine. If we had a larger sample size, we may be able to observe enough individuals who did not gain weight or even had weight loss over their lifetime. We were also limited in examination of subgroups, particularly stratifications by sex given the small number of female cases available for analysis. However, we present the stratifications by sex and study in Supplementary Material, as these stratifications are of interest due to the sex disparity in EA/GCA incidence and possible between-study heterogeneity. Although we did not run competing risk analyses, our primary aim was to estimate cause-specific relative risk, which does not require independence of the outcome and competing events to obtain valid relative risk estimates (Prentice et al, 1978). In addition, the current study includes older USA participants only, which reduces the generalisability of results.
In summary, this analysis of two large, mature US cohorts provide evidence that early adulthood overweight and obesity are strong risk factors for EA and GCA, and that large weight gains over the life course confer high risks for these malignancies. These results indicate that weight gain during adulthood should be avoided to reduce the risk of EA/GCA, and underscores the potential of weight control programs for reducing the incidence of these highly lethal cancers.
Change history
28 March 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Abrams P, Levitt Katz LE (2011) Metabolic effects of obesity causing disease in childhood. Curr Opin Endocrinol Diabetes Obes 18 (1): 23–27.
Adams KF, Leitzmann MF, Ballard-Barbash R, Albanes D, Harris TB, Hollenbeck A, Kipnis V (2014) Body mass and weight change in adults in relation to mortality risk. Am J Epidemiol 179 (2): 135–144.
Adegboye AR, Andersen LB, Froberg K, Sardinha LB, Heitmann BL (2010) Linking definition of childhood and adolescent obesity to current health outcomes. Int J Pediatr Obes 5 (2): 130–142.
Cheng KK, Sharp L, McKinney PA, Logan RF, Chilvers CE, Cook-Mozaffari P, Ahmed A, Day NE (2000) A case-control study of oesophageal adenocarcinoma in women: a preventable disease. Br J Cancer 83 (1): 127–132.
Chow WH, Blot WJ, Vaughan TL, Risch HA, Gammon MD, Stanford JL, Dubrow R, Schoenberg JB, Mayne ST, Farrow DC, Ahsan H, West AB, Rotterdam H, Niwa S, Fraumeni JF Jr (1998) Body mass index and risk of adenocarcinomas of the esophagus and gastric cardia. J Natl Cancer Inst 90 (2): 150–155.
Cook MB, Freedman ND, Gamborg M, Sorensen TI, Baker JL (2015) Childhood body mass index in relation to future risk of oesophageal adenocarcinoma. Br J Cancer 112 (3): 601–607.
Corley DA, Kubo A (2004) Influence of site classification on cancer incidence rates: an analysis of gastric cardia carcinomas. J Natl Cancer Inst 96 (18): 1383–1387.
Dahl AK, Hassing LB, Fransson EI, Pedersen NL (2010) Agreement between self-reported and measured height, weight and body mass index in old age—a longitudinal study with 20 years of follow-up. Age Ageing 39 (4): 445–451.
Dahl AK, Reynolds CA (2013) Accuracy of recalled body weight—a study with 20-years of follow-up. Obesity (Silver Spring) 21 (6): 1293–1298.
de Mutsert R, Sun Q, Willett WC, Hu FB, van Dam RM (2014) Overweight in early adulthood, adult weight change, and risk of type 2 diabetes, cardiovascular diseases, and certain cancers in men: a cohort study. Am J Epidemiol 179 (11): 1353–1365.
Devesa SS, Blot WJ, Fraumeni JF Jr (1998) Changing patterns in the incidence of esophageal and gastric carcinoma in the United States. Cancer 83 (10): 2049–2053.
Falk GW (2009) Risk factors for esophageal cancer development. Surg Oncol Clin N Am 18 (3): 469–485.
Finkelstein EA, Khavjou OA, Thompson H, Trogdon JG, Pan L, Sherry B, Dietz W (2012) Obesity and severe obesity forecasts through 2030. Am J Prev Med 42 (6): 563–570.
Flegal KM, Shepherd JA, Looker AC, Graubard BI, Borrud LG, Ogden CL, Harris TB, Everhart JE, Schenker N (2009) Comparisons of percentage body fat, body mass index, waist circumference, and waist-stature ratio in adults. Am J Clin Nutr 89 (2): 500–508.
Hoyo C, Cook MB, Kamangar F, Freedman ND, Whiteman DC, Bernstein L, Brown LM, Risch HA, Ye W, Sharp L, Wu AH, Ward MH, Casson AG, Murray LJ, Corley DA, Nyren O, Pandeya N, Vaughan TL, Chow WH, Gammon MD (2012) Body mass index in relation to oesophageal and oesophagogastric junction adenocarcinomas: a pooled analysis from the International BEACON Consortium. Int J Epidemiol 41 (6): 1706–1718.
Jones BL, Nagin DS (2007) Advances in group-based trajectory modeling and an SAS procedure for estimating them. Socio Meth Res 35 (4): 542–571.
Kleinbaum D (2002) Logistic Regression: a Self-Learning Text 2nd edn, New York: Springer.
Koebnick C, Getahun D, Smith N, Porter AH, Der-Sarkissian JK, Jacobsen SJ (2011) Extreme childhood obesity is associated with increased risk for gastroesophageal reflux disease in a large population-based study. Int J Pediatr Obes 6 (2-2): e257–e263.
Kramer BS, Gohagan J, Prorok PC, Smart C (1993) A National Cancer Institute sponsored screening trial for prostatic, lung, colorectal, and ovarian cancers. Cancer 71 (2 Suppl): 589–593.
Lagergren J, Bergstrom R, Nyren O (1999) Association between body mass and adenocarcinoma of the esophagus and gastric cardia. Ann Intern Med 130 (11): 883–890.
Macfarlane GJ, de Silva V, Jones GT (2011) The relationship between body mass index across the life course and knee pain in adulthood: results from the 1958 birth cohort study. Rheumatology (Oxford) 50 (12): 2251–2256.
Malaty HM, Fraley JK, Abudayyeh S, Fairly KW, Javed US, Aboul-Fotouh H, Mattek N, Gilger MA (2009) Obesity and gastroesophageal reflux disease and gastroesophageal reflux symptoms in children. Clin Exp Gastroenterol 2: 31–36.
Merry AH, Schouten LJ, Goldbohm RA, van den Brandt PA (2007) Body mass index, height and risk of adenocarcinoma of the oesophagus and gastric cardia: a prospective cohort study. Gut 56 (11): 1503–1511.
Michaud D, Midthune D, Hermansen S, Leitzmann M, Harlan LC, Kipnis V, Schatzkin A (2005) Comparison of cancer registry case ascertainment with SEER estimates and self-reporting in a subset of the NIH-AARP Diet and Health Study. J Registry Manag 32 (2): 70–75.
Oldham PD (1962) A note on the analysis of repeated measurements of the same subjects. J Chronic Dis 15: 969–977.
Pashankar DS, Corbin Z, Shah SK, Caprio S (2009) Increased prevalence of gastroesophageal reflux symptoms in obese children evaluated in an academic medical center. J Clin Gastroenterol 43 (5): 410–413.
Prentice RL, Kalbfleisch JD, Peterson AV Jr, Flournoy N, Farewell VT, Breslow NE (1978) The analysis of failure times in the presence of competing risks. Biometrics 34 (4): 541–554.
Reid BJ, Li X, Galipeau PC, Vaughan TL (2010) Barrett’s oesophagus and oesophageal adenocarcinoma: time for a new synthesis. Nat Rev Cancer 10 (2): 87–101.
Rothman KJ, Greenland S, Lash TL (2008) Modern Epidemiology 3rd edn, Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins.
Ruigomez A, Lundborg P, Johansson S, Wallander MA, Garcia Rodriguez LA (2010) Follow-up of a cohort of children and adolescents with gastro-esophageal reflux disease who were free of reflux esophagitis at initial diagnosis. Scand J Gastroenterol 45 (7-8): 814–821.
Ryan AM, Duong M, Healy L, Ryan SA, Parekh N, Reynolds JV, Power DG (2011) Obesity, metabolic syndrome and esophageal adenocarcinoma: epidemiology, etiology and new targets. Cancer Epidemiol 35 (4): 309–319.
Schatzkin A, Subar AF, Thompson FE, Harlan LC, Tangrea J, Hollenbeck AR, Hurwitz PE, Coyle L, Schussler N, Michaud DS, Freedman LS, Brown CC, Midthune D, Kipnis V (2001) Design and serendipity in establishing a large cohort with wide dietary intake distributions : the National Institutes of Health-American Association of Retired Persons Diet and Health Study. Am J Epidemiol 154 (12): 1119–1125.
Sharma P, McQuaid K, Dent J, Fennerty MB, Sampliner R, Spechler S, Cameron A, Corley D, Falk G, Goldblum J, Hunter J, Jankowski J, Lundell L, Reid B, Shaheen NJ, Sonnenberg A, Wang K, Weinstein W (2004) A critical review of the diagnosis and management of Barrett’s esophagus: the AGA Chicago Workshop. Gastroenterology 127 (1): 310–330.
Simard EP, Ward EM, Siegel R, Jemal A (2012) Cancers with increasing incidence trends in the United States: 1999 through 2008. CA Cancer J Clin 62 (2): 118–128.
Singh AS, Mulder C, Twisk JW, van Mechelen W, Chinapaw MJ (2008) Tracking of childhood overweight into adulthood: a systematic review of the literature. Obes Rev 9 (5): 474–488.
Singh S, Sharma AN, Murad MH, Buttar NS, El-Serag HB, Katzka DA, Iyer PG (2013) Central adiposity is associated with increased risk of esophageal inflammation, metaplasia, and adenocarcinoma: a systematic review and meta-analysis. Clin Gastroenterol Hepatol 11 (11): 1399–1412 e7.
Song M, Willett WC, Hu FB, Spiegelman D, Must A, Wu K, Chan AT, Giovannucci EL (2015) Trajectory of body shape across the lifespan and cancer risk. Int J Cancer 138 (10): 2383–2395.
Spencer EA, Appleby PN, Davey GK, Key TJ (2002) Validity of self-reported height and weight in 4808 EPIC-Oxford participants. Public Health Nutr 5 (4): 561–565.
Tu YK, Gilthorpe MS (2007) Revisiting the relation between change and initial value: a review and evaluation. Stat Med 26 (2): 443–457.
WHO (2000) Obesity: preventing and managing the global epidemic. Report of a WHO consultation. World Health Organ Tech Rep Ser 894: i-xii, 1–253.
Wijnhoven BP, Siersema PD, Hop WC, van Dekken H, Tilanus HW (1999) Adenocarcinomas of the distal oesophagus and gastric cardia are one clinical entity. Rotterdam Oesophageal Tumour Study Group. Br J Surg 86 (4): 529–535.
Wild CP, Hardie LJ (2003) Reflux, Barrett's oesophagus and adenocarcinoma: burning questions. Nat Rev Cancer 3 (9): 676–684.
Wu AH, Wan P, Bernstein L (2001) A multiethnic population-based study of smoking, alcohol and body size and risk of adenocarcinomas of the stomach and esophagus (United States). Cancer Causes Control 12 (8): 721–732.
Acknowledgements
This research was supported (in part) by the Intramural Research Programme of the NIH, National Cancer Institute. Cancer incidence data from the Atlanta metropolitan area were collected by the Georgia Center for Cancer Statistics, Department of Epidemiology, Rollins School of Public Health, Emory University, Atlanta, Georgia. Cancer incidence data from California were collected by the California Cancer Registry, California Department of Public Health’s Cancer Surveillance and Research Branch, Sacramento, California. Cancer incidence data from the Detroit metropolitan area were collected by the Michigan Cancer Surveillance Programme, Community Health Administration, Lansing, Michigan. The Florida cancer incidence data used in this report were collected by the Florida Cancer Data System (Miami, Florida) under contract with the Florida Department of Health, Tallahassee, Florida. Cancer incidence data from Louisiana were collected by the Louisiana Tumor Registry, Louisiana State University Health Sciences Center School of Public Health, New Orleans, Louisiana. Cancer incidence data from New Jersey were collected by the New Jersey State Cancer Registry, The Rutgers Cancer Institute of New Jersey, New Brunswick, New Jersey. Cancer incidence data from North Carolina were collected by the North Carolina Central Cancer Registry, Raleigh, North Carolina. Cancer incidence data from Pennsylvania were supplied by the Division of Health Statistics and Research, Pennsylvania Department of Health, Harrisburg, Pennsylvania. The Pennsylvania Department of Health specifically disclaims responsibility for any analyses, interpretations or conclusions. Cancer incidence data from Arizona were collected by the Arizona Cancer Registry, Division of Public Health Services, Arizona Department of Health Services, Phoenix, Arizona. Cancer incidence data from Texas were collected by the Texas Cancer Registry, Cancer Epidemiology and Surveillance Branch, Texas Department of State Health Services, Austin, Texas. Cancer incidence data from Nevada were collected by the Nevada Central Cancer Registry, Division of Public and Behavioral Health, State of Nevada Department of Health and Human Services, Carson City, Nevada. We are indebted to the participants in the NIH-AARP Diet and Health Study for their outstanding cooperation. We also thank Sigurd Hermansen and Kerry Grace Morrissey from Westat for study outcomes ascertainment and management, and Leslie Carroll at Information Management Services for data support and analysis.
Disclaimer
The views expressed herein are solely those of the authors and do not necessarily reflect those of the FCDC or FDOH.
Author contributions
JLP participated in the conception, design, and analysis of the study; drafted the manuscript; and approved the final manuscript. SPK participated in the conception, design, and analysis of the study; and provided critical revision of the manuscript for important intellectual content; and approved the final manuscript. LML participated in the design of the study; provided critical revision of the manuscript for important intellectual content; and approved the final manuscript. NDF participated in the design of the study; provided critical revision of the manuscript for important intellectual content; and approved the final manuscript. BIG participated in the conception, design, and analysis of the study; and provided critical revision of the manuscript for important intellectual content; and approved the final manuscript. MBC participated in the conception, design, and analysis of the study; assisted in drafting the manuscript and providing revisions of important intellectual content; and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Petrick, J., Kelly, S., Liao, L. et al. Body weight trajectories and risk of oesophageal and gastric cardia adenocarcinomas: a pooled analysis of NIH-AARP and PLCO Studies. Br J Cancer 116, 951–959 (2017). https://doi.org/10.1038/bjc.2017.29
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.29
Keywords
This article is cited by
-
Metabolic health's central role in chronic kidney disease progression: a 20-year study of obesity-metabolic phenotype transitions
Scientific Reports (2024)
-
Association between circadian physical activity trajectories and incident type 2 diabetes in the UK Biobank
Scientific Reports (2024)
-
Self-Reported Early and Later Life Weight and the Risk of All-Cause Mortality in Older Adults
The Journal of nutrition, health and aging (2023)
-
Associations of genetic risk, BMI trajectories, and the risk of non-small cell lung cancer: a population-based cohort study
BMC Medicine (2022)
-
Associations of specific-age and decade recall body mass index trajectories with obesity-related cancer
BMC Cancer (2021)
