
Abstract
Background:
Neuroendocrine tumours (NETs) are rare in children and limited data are available. We aimed to specify tumour and patient characteristics and to investigate the role of genetic predisposition in the aetiology of paediatric NETs.
Methods:
Using the Dutch Pathology Registry PALGA, we collected patient- and tumour data of paediatric NETs in the Netherlands between 1991 and 2013 (N=483).
Results:
The incidence of paediatric NETs in the Netherlands is 5.40 per one million per year. The majority of NETs were appendiceal tumours (N=441;91.3%). Additional surgery in appendiceal NETs was indicated in 89 patients, but performed in only 27 of these patients. Four out of five patients with pancreatic NETs were diagnosed with Von Hippel–Lindau disease (N=2) and Multiple Endocrine Neoplasia type 1 (N=2). In one patient with an appendiceal NET Familial Adenomatous Polyposis was diagnosed. On the basis of second primary tumours or other additional diagnoses, involvement of genetic predisposition was suggestive in several others.
Conclusions:
We identified a significant number of patients with a confirmed or suspected tumour predisposition syndrome and show that paediatric pancreatic NETs in particular are associated with genetic syndromes. In addition, we conclude that treatment guidelines for appendiceal paediatric NETs need revision and improved implementation.
Similar content being viewed by others

Main
A neuroendocrine tumour (NET) is a rare tumour with an estimated incidence of 5.25 per 100 000 in adults (Yao et al, 2008; Lawrence et al, 2011). The exact incidence of NETs in children is not known, but a small study reported an incidence of 1.14 per one million children (Parkes et al, 1993). In children, little research has been performed on NETs with only small case-series of 2 to 36 patients reported (Kulkarni and Sergi, 2013).
Neuroendocrine tumours in children seem to differ from NETs in adults. In adults, the most common locations of NETs are the lungs and the gastrointestinal tract, specifically the rectum and small intestine (Yao et al, 2008). In children, NETs are most commonly found in the appendix (87.5% Fernandez et al, 2015), whereas in adults the incidence of NETs in the appendix is relatively low (3% of all NETs; Yao et al, 2008). Appendiceal NETs in children are indolent tumours which rarely metastasise and have an excellent overall survival close to 100% (Kulkarni and Sergi, 2013). Survival in adults depends on the localisation of the NET and disease stage at diagnosis (Yao et al, 2008). The 5-year survival rate of localised appendiceal NETs is 88% (Yao et al, 2008), which is less than the estimated survival in children.
Genetic predisposition may have a role in the development of NETs. These tumours have been reported as part of various hereditary cancer syndromes as shown in Table 1, and familial occurrence of NETs has also been reported in the absence of these syndromes (Hemminki and Li, 2001). In addition, adults diagnosed with a NET have an increased risk of developing second primary malignancies (Babovic-Vuksanovic et al, 1999). These characteristics are suggestive for hitherto unrecognised genetic predisposing factors involved in the development of NETs in adults.
On the basis of the findings in adult studies, we suspect genetic tumour predisposition syndromes might have a role in the aetiology of NETs in children as well. To support this hypothesis we studied the occurrence of second primary malignancies and the presence of additional features or diagnoses in children with a NET.
Here we describe a large study of NETs in 483 children in the Netherlands using PALGA, the nationwide pathology database. The first aim of this study was to provide an overview of patient and tumour characteristics in children with a NET. The collected data also enabled us to study the adherence to surgical guidelines in children with appendiceal NETs. Second, we aimed to find evidence to support our hypothesis that genetic tumour predisposition is a contributing factor in the aetiology of NETs. To our knowledge, this is the largest case-series described in the literature so far.
Materials and methods
We retrospectively collected our data from PALGA, the nationwide network and registry of histopathology and cytopathology (Casparie et al, 2007) which encompasses all pathology reports from all hospitals in the Netherlands since 1991. To obtain data from all children in the Netherlands with a NET, we performed a search with the search terms ‘carcinoid’ OR ‘neuroendocrine tumour’ with limitations to the ages 0–18. This search resulted in data from 505 children who were diagnosed with a NET between 1991 and 2013. In the Bosman et al (2010) and in our national guidelines no diagnostic requirements for the diagnosis of a NET are defined. Diagnosis was based on HE staining in unequivocal cases and additional immunohistochemical staining if deemed necessary by the pathologist, as is in daily practice. Data were processed de-identified and contained 1105 excerpts in total. Information about age at diagnosis, gender, localisation of the tumour, tumour size, presence of metastasis, surgical treatment and presence of additional features or diagnoses was extracted. The file also included the data about other conditions for which a pathologist was consulted that occurred prior to or in follow-up of the NET. Many reports included the data on the medical history of the child, which could shed light on conditions for which examination by a pathologist was not necessary.
A total of 22 patients were excluded from the final analysis; six because data were incomplete and 16 due to another diagnosis. Patients in the latter group did not have a NET, but a different tumour with neuroendocrine characteristics or a different type of tumour with a NET only in the initial differential diagnosis.
Patient characteristics were compared using crosstabs in conjunction with a standard Pearson chi-square test. Statistical significance was considered to exist if two-sided P-values were below 0.01. The incidence of NETs was calculated by dividing the number of children with a NET by the total Dutch childhood population for each of the years between 2009 to 2013 (available at statline.cbs.nl). The average incidence is based on these 5 years. Statistical analysis was carried out using the SPSS statistical package (IBM Corp, 2013). Both the privacy committee and the scientific committee of PALGA approved the study design. Because of the strict anonymity, no additional ethical approval was required.
Results
Patient and tumour characteristics
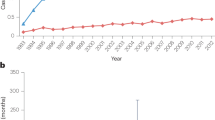
A total of 483 patients (174 males and 309 females) were included in the analysis. The mean follow-up of patients was 143 months (median 146 months, range 2–275 months). From 1991 to 2013 on average 21 children (range 14–26) were diagnosed with a NET per year in the Netherlands, which averages an incidence rate of 5.40 per million children per year. The median age of patients at the time of diagnosis was 15 years (range 1–18 years) and NETs were more frequently seen in girls. We found a male : female ratio of 1 : 1.78. We observed no difference in age at diagnosis by sex or by primary tumour localisation. Details regarding age at diagnosis and the distribution of tumour localisation are presented in Table 2.
Appendiceal NETs were observed most frequently (441 patients, 91.3%), followed by bronchopulmonary tumours (25 patients, 5.2%). Two patients presented with a NET localised in another (benign) tumour: a NET in a mature cystic teratoma and a NET in a mature sacrococcygeal teratoma. In one patient with liver metastasis of a NET, the primary tumour could not be identified. The localisations of the primary tumours in the patients varied significantly by sex, with increased incidence of appendiceal NETs in females (95.1% vs 84.5%, P<0.001) and increased incidence of bronchopulmonary NETs in male patients (10.3% vs 2.3%, P<0.001). The distribution of gender related to tumour location is shown in Table 3.
Metastatic disease
At the time of diagnosis, the majority of patients did not have metastatic disease (N=467, 96.7%). In 13 patients (2.7%) locoregional lymph node metastasis was present. In three patients (0.6%), distant metastasis to the liver was found. Of these patients one had a primary NET in the stomach and the second a NET in a mature cystic teratoma. In the third patient with distant metastasis the primary tumour was not identified after an extensive search including multiple stomach biopsies and an appendectomy. Appendiceal NETs metastasise significantly less than NETs in other locations (P<0.001). Extended patient and tumour characteristics of patients with metastatic disease are described in Supplementary Table 1.
Appendiceal tumours
We found the appendix to be the most common location of a NET in children (n=441, 91.3%). Histopathological data of the appendiceal NETs are presented in Table 4. Four patients had multiple NETs in the appendix, with a maximum of four NETs in one appendix (Table 5). All but two patients presented with clinical symptoms of an acute abdomen, suggestive of appendicitis. In the remaining two patients, the appendiceal NETs were incidentally found during surgery for another indication: a patient with Crohn’s disease underwent an ileocecal resection and a patient with Familial adenomatous polyposis (FAP) underwent a proctocolectomy. In 403 patients, an appendectomy was performed without additional surgery. In 33 patients an additional hemicolectomy or ileocecal resection was performed.
Genetic predisposition
Genetic tumour predisposition can be suspected when a child has multiple primary tumours, multiple NETs or specific additional features or diagnoses that could indicate genetic predisposition. A total of 18 patients in our cohort matched these criteria (Table 5). In six patients (1.2%) a second primary malignancy was found. Two of these patients had Von Hippel–Lindau disease (VHL) and one was clinically diagnosed with Multiple Endocrine Neoplasia syndrome type 1 (MEN-1), although from the PALGA report we learned that a causative mutation had not yet been identified. Three patients with an appendiceal NET (0.6%) had additional premalignant lesions. One patient had multiple adenomatous polyps of low to intermediate grade dysplasia in the colon and in this patient the tumour predisposition syndrome FAP was diagnosed. The other patient had CIN2 moderate dysplasia of the cervix at a remarkably young age of 17 years. The third patient had a dysplastic naevus, also at a very young age of 10 years.
Five patients (1.0%) presented with multiple NETs in the same organ. Four had multiple appendiceal NETs and one patient had multiple pancreatic NETs. This latter patient also had a MEN-1 diagnosis. Four other patients had additional features or diagnoses that have been described in tumour predisposition syndromes before, that is pilomatrixomas, multiple angiolipomas and an osteoma. We found a high incidence of Crohn’s disease or strongly suspected inflammatory bowel disease in 9 patients (1.9%, 2 confirmed, 7 patients suspected). An overview of all additional diagnoses found in our cohort is available in Supplementary Table 2.
Discussion
To our knowledge, this is the largest case-series of NETs in children described in literature and we were able to confirm previously reported characteristics of these tumours. In addition, our results suggest that genetic tumour predisposition has an important role in the aetiology of NETs in children, as we found proven and strongly suspected genetic tumour predisposition syndromes in 18 children.
The incidence of 5.40 per one million children per year in the Netherlands is higher than the estimated incidence of 1.14 per million children (Parkes et al, 1993). The incidence we report is probably more realistic since we conducted a nationwide study. In line with previous studies, we found that the appendix is by far the most common site for NETs in children (Neves et al, 2006). Tumour localisation varied significantly between sexes with females developing significantly more appendiceal NETs then males. This may be explained by the increased lifetime risk for a female to undergo an appendectomy (23.1% vs 12.0% for males; Addiss et al, 1990) with consequently more incidental findings of appendiceal NETs. It is unknown whether this explanation is also applicable to the childhood population. Compared with females, males develop more bronchopulmonary NETs. Strikingly, in adult studies, females develop more bronchopulmonary NETs (Modlin et al, 2003; Yao et al, 2008). We do not have an explanation for the predilection of bronchopulmonary NETs in males in our cohort. However, the number of pulmonary tumours we found is relatively small and this finding needs confirmation.
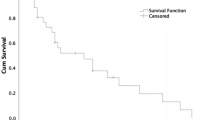
A NET in children is reported to have a relatively indolent disease course (Spunt et al, 2000; Kulkarni and Sergi, 2013). Data about survival are lacking in our study due to the study design, however only a few patients developed locoregional lymph node metastases (N=13) or distant metastases (N=3). NETs in the stomach, colon, lungs and NETs localised in different tumours seem to metastasise more often (50, 20, 16 and 50%, respectively). Because these localisations are rare in children these percentages are based on small numbers and therefore the strength of this observation may be questionable. However, stricter surveillance in children with these tumour localisations seems warranted.
Exact guidelines for the surgical treatment of appendiceal NETs in children are not available. In adult patients, a right-sided hemicolectomy or an ileocecal resection is advised when histopathological risk factors are present, according to the ENETS consensus treatment guidelines for neuroendocrine tumours of the appendix (Pape et al, 2016). The ENETS consensus guidelines state that additional surgery is indicated when one of the following criteria is present: tumour size >2 cm, invasion of the mesoappendix, localisation of the tumour at the base of the appendix or an incomplete initial resection of the tumour (Pape et al, 2016). On the basis of these criteria additional surgery was indicated in 89 patients with appendiceal NETs, but performed in only 27 of these patients. Six patients had additional surgery, although there was no strict indication according to these guidelines. It is noteworthy that these treatment guidelines were not available in the early nineties, so they are not applicable for our entire cohort. No clear difference was seen between the group of patients in whom additional surgery was performed, compared with the group of patients in whom additional surgery was not performed. On the other hand, six patients did have additional surgery despite the fact that it was not indicated based on the tumour characteristics. The role of additional surgery in the management of patients with appendiceal NETs is controversial, particularly in children and adolescents, as NETs in these patients tend to behave less aggressively (Safioleas et al, 2005). Indeed, none of the 62 patients who did not undergo additional surgery developed histological proven metastatic disease during follow-up. Perhaps clinicians consider these guidelines to extreme for these tumours in children and therefore they opt for a less invasive treatment. The development of a guideline for surgical treatment in children with appendiceal NETs may help find more consensus on this subject.
We found a significant number of patients (N=18, 3.7% Table 5) in whom a genetic tumour predisposition syndrome was diagnosed or strongly suspected. Genetic predisposition can be suspected based on several factors, like for instance the presence of multiple primary tumours in a patient (Moppett et al, 2001) and cancer in combination with specific features that fit one of the known tumour predisposition syndromes, such as specific skin lesions (e.g., café-au-lait macules in neurofibromatosis) or congenital anomalies (Merks et al, 2008). Finally, a family history of cancer could be indicative of genetic predisposition. Unfortunately, data about family history were unavailable in our study.
A total of six patients developed a second primary malignancy (SPM) during the period of observation (1.2%). None of these malignancies was treatment related since none of these children received chemotherapy or radiotherapy. Therefore, these children had an indication for genetic counselling and genetic testing (Moppett et al, 2001). In three of these children, we indeed found evidence for a known cancer predisposing syndrome (Table 5). The two tumour predisposition syndromes VHL and MEN-1 occurred in a much higher incidence in our cohort than in the general population (1 : 242 vs 1 : 36 000 (Lonser et al, 2003) and 1 : 242 vs 1 : 50 000 (Brandi et al, 2001), respectively; P<0.001 for both comparisons). All of these patients had pancreatic NETs, which suggests pancreatic NETs in particular are highly associated with tumour predisposition syndromes. We could not confirm that a genetic predisposition syndrome was present in the other three children with multiple malignancies, which does not necessarily mean that such a diagnosis has not been made. In particular, genetic tumour predisposition is likely to have a role in the patient with an adenoid cystic breast carcinoma at age 17, since this is a tumour that is very unusual in childhood. Mean age at diagnosis of this tumour is 63 years (Ghabach et al, 2010) and to our knowledge, no case reports of this tumour in children have been described.
In studies on SPMs in adult patients with gastrointestinal NETs, the incidence of SPMs ranges from 12–46% (Habal et al, 2000). In children, only one retrospective study was performed and no SPMs were identified (Fernandez et al, 2015). However, this study was rather small and contained a cohort of only 32 patients with a short median follow-up of 84 months. Even though our follow-up was relatively short in many of the patients (median of 143 months, range 2–275) we identified 6 patients with a SPM. If follow-up would be extended, it is possible that more SPMs at relatively young age will be identified.
We have identified one child with FAP and several children with Crohn’s disease in our cohort. Neuroendocrine tumours in FAP and Crohn’s disease have been described before (July et al, 1999; West et al, 2007; Pratico et al, 2013). We found an incidence of Crohn’s disease or strongly suspected inflammatory bowel disease of 0.4–1.9% (400–1900 per 100 000) which is significantly higher than the incidence of Crohn’s disease in the general paediatric population (6.6 per 100 000; P<0.001; Adamiak et al, 2013). The most important explanation for the association between Crohn’s disease and NETs is probably the higher number of colectomies performed in children with these conditions, which can result in the incidental finding of a NET (Derikx et al, 2016). The hypothesis that inflammation causes hyperstimulation of endocrine cells leading to hyperplasia and neoplasia remains to be proven (West et al, 2007). A third explanation might be that genetic predisposing factors for FAP and Crohn also predispose for developing NETs.
The other remarkable findings shown in Table 5 are features that are not previously reported in patients with NETs, but some are associated with tumour predisposition syndromes in which NETs have been described. For example, pilomatrixomas and osteomas have been described in FAP (Bisgaard and Bulow, 2006; Trufant et al, 2012). Since these are all rare findings in children, the combination with childhood cancer may suggest the presence of a genetic predisposition syndrome. Possibly even more patients have interesting additional diagnoses, but using PALGA as our main source of information we missed the patients with additional diagnoses of which no biopsy was taken or tissue removed.
Conclusion
We identified several patients with a confirmed or suspected tumour predisposition syndrome and showed that paediatric pancreatic NETs in particular are associated with tumour predisposition syndromes, since 4 out of 5 patients with a pancreatic NET had such a syndrome. The presence of a pancreatic NET in a child should therefore warrant genetic counselling, genetic testing and surveillance advises in line with the results of genetic testing. Studies including exome- or genome-wide genetic testing are needed to further identify the role of genetic predisposition in the aetiology of NETs.
In addition, we found that the current treatment guidelines need revision and improved implementation in light of the much better prognosis of appendiceal NETs in children compared with adults.
Change history
17 January 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Adamiak T, Walkiewicz-Jedrzejczak D, Fish D, Brown C, Tung J, Khan K, Faubion W Jr ., Park R, Heikenen J, Yaffee M, Rivera-Bennett MT, Wiedkamp M, Stephens M, Noel R, Nugent M, Nebel J, Simpson P, Kappelman MD, Kugathasan S (2013) Incidence, clinical characteristics, and natural history of pediatric IBD in Wisconsin: a population-based epidemiological study. Inflamm Bowel Dis 19 (6): 1218–1223.
Addiss DG, Shaffer N, Fowler BS, Tauxe RV (1990) The epidemiology of appendicitis and appendectomy in the United States. Am J Epidemiol 132 (5): 910–925.
Anlauf M, Garbrecht N, Bauersfeld J, Schmitt A, Henopp T, Komminoth P, Heitz PU, Perren A, Kloppel G (2007) Hereditary neuroendocrine tumors of the gastroenteropancreatic system. Virchows Arch 451 (Suppl 1): S29–S38.
Babovic-Vuksanovic D, Constantinou CL, Rubin J, Rowland CM, Schaid DJ, Karnes PS (1999) Familial occurrence of carcinoid tumors and association with other malignant neoplasms. Cancer Epidemiol Biomarkers Prev 8 (8): 715–719.
Bisgaard ML, Bulow S (2006) Familial adenomatous polyposis (FAP): genotype correlation to FAP phenotype with osteomas and sebaceous cysts. Am J Med Genet A 140 (3): 200–204.
Blansfield JA, Choyke L, Morita SY, Choyke PL, Pingpank JF, Alexander HR, Seidel G, Shutack Y, Yuldasheva N, Eugeni M, Bartlett DL, Glenn GM, Middelton L, Linehan WM, Libutti SK (2007) Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs). Surgery 142 (6): 814–818, discussion 818 e1-2.
Bosman FT, Carneiro F, Hruban RH, Theise ND (2010) WHO Classification of Tumours of the Digestive System, 4th edn, WHO press: Geneva, Switzerland.
Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, Conte-Devolx B, Falchetti A, Gheri RG, Libroia A, Lips CJ, Lombardi G, Mannelli M, Pacini F, Ponder BA, Raue F, Skogseid B, Tamburrano G, Thakker RV, Thompson NW, Tomassetti P, Tonelli F, Wells SA Jr ., Marx SJ (2001) Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 86 (12): 5658–5671.
Camp ER, Hochwald SN, Liu C (2004) FAP with concurrent duodenal adenomatous polyposis and carcinoid tumor. J Surg Oncol 87 (4): 187–190.
Casparie M, Tiebosch AT, Burger G, Blauwgeers H, van de Pol A, van Krieken JH, Meijer GA (2007) Pathology databanking and biobanking in The Netherlands, a central role for PALGA, the nationwide histopathology and cytopathology data network and archive. Cell Oncol 29 (1): 19–24.
Derikx LA, Vierdag WM, Kievit W, Bosch S, Hoentjen F, Nagtegaal ID (2016) Is the prevalence of colonic neuroendocrine tumors increased in patients with inflammatory bowel disease? Int J Cancer 139 (3): 535–542.
Fernandez KS, Aldrink JH, Ranalli M, Ruymann FB, Caniano DA (2015) Carcinoid tumors in children and adolescents: risk for second malignancies. J Pediatr Hematol Oncol 37 (2): 150–153.
Ghabach B, Anderson WF, Curtis RE, Huycke MM, Lavigne JA, Dores GM (2010) Adenoid cystic carcinoma of the breast in the United States (1977 to 2006): a population-based cohort study. Breast Cancer Res 12 (4): R54.
Goudet P, Bonithon-Kopp C, Murat A, Ruszniewski P, Niccoli P, Menegaux F, Chabrier G, Borson-Chazot F, Tabarin A, Bouchard P, Cadiot G, Beckers A, Guilhem I, Chabre O, Caron P, Du Boullay H, Verges B, Cardot-Bauters C (2011) Gender-related differences in MEN1 lesion occurrence and diagnosis: a cohort study of 734 cases from the Groupe d'etude des Tumeurs Endocrines. Eur J Endocrinol 165 (1): 97–105.
Goudet P, Murat A, Cardot-Bauters C, Emy P, Baudin E, du Boullay Choplin H, Chapuis Y, Kraimps JL, Sadoul JL, Tabarin A, Verges B, Carnaille B, Niccoli-Sire P, Costa A, Calender A, network GTE (2009) Thymic neuroendocrine tumors in multiple endocrine neoplasia type 1: a comparative study on 21 cases among a series of 761 MEN1 from the GTE (Groupe des Tumeurs Endocrines). World J Surg 33 (6): 1197–1207.
Habal N, Sims C, Bilchik AJ (2000) Gastrointestinal carcinoid tumors and second primary malignancies. J Surg Oncol 75 (4): 310–316.
Hammel PR, Vilgrain V, Terris B, Penfornis A, Sauvanet A, Correas JM, Chauveau D, Balian A, Beigelman C, O'Toole D, Bernades P, Ruszniewski P, Richard S (2000) Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francophone d'Etude de la Maladie de von Hippel-Lindau. Gastroenterology 119 (4): 1087–1095.
Hemminki K, Li X (2001) Familial carcinoid tumors and subsequent cancers: a nation-wide epidemiologic study from Sweden. Int J Cancer 94 (3): 444–448.
IBM Corp. Released (2013) IBM SPSS Statistics for Windows, Version 22.0. IBM Corp: Armonk, NY, USA.
July LV, Northcott KA, Yoshida EM, Carr DM, Owen DA (1999) Coexisting carcinoid tumors in familial adenomatous polyposis-associated upper intestinal adenomas. Am J Gastroenterol 94 (4): 1091–1094.
Kulkarni KP, Sergi C (2013) Appendix carcinoids in childhood: long-term experience at a single institution in Western Canada and systematic review. Pediatr Int 55 (2): 157–162.
Lawrence B, Gustafsson BI, Chan A, Svejda B, Kidd M, Modlin IM (2011) The epidemiology of gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am 40 (1): 1–18, vii.
Lodish MB, Stratakis CA (2010) Endocrine tumours in neurofibromatosis type 1, tuberous sclerosis and related syndromes. Best Pract Res Clin Endocrinol Metab 24 (3): 439–449.
Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH (2003) von Hippel-Lindau disease. Lancet 361 (9374): 2059–2067.
Merks JH, Ozgen HM, Koster J, Zwinderman AH, Caron HN, Hennekam RC (2008) Prevalence and patterns of morphological abnormalities in patients with childhood cancer. JAMA 299 (1): 61–69.
Miquel C, Sabourin JC, Elias D, Grandjouan S, Viguier J, Ducreux M, Duvillard P, Praz F (2004) An appendix carcinoid tumor in a patient with hereditary nonpolyposis colorectal cancer. Hum Pathol 35 (12): 1564–1567.
Modlin IM, Lye KD, Kidd M (2003) A 5-decade analysis of 13,715 carcinoid tumors. Cancer 97 (4): 934–959.
Moppett J, Oakhill A, Duncan AW (2001) Second malignancies in children: the usual suspects? Eur J Radiol 38 (3): 235–248.
Neves GR, Chapchap P, Sredni ST, Viana CR, Mendes WL (2006) Childhood carcinoid tumors: description of a case series in a Brazilian cancer center. Sao Paulo Med J 124 (1): 21–25.
Pape UF, Niederle B, Costa F, Gross D, Kelestimur F, Kianmanesh R, Knigge U, Oberg K, Pavel M, Perren A, Toumpanakis C, O'Connor J, Krenning E, Reed N, O'Toole D (2016) ENETS Consensus Guidelines for Neuroendocrine Neoplasms of the Appendix (Excluding Goblet Cell Carcinomas). Neuroendocrinology 103 (2): 144–152.
Parkes S, Muir K, Al Sheyyab M (1993) Carcinoid tumors of the appendix in children 1957–1986: incidence, treatment and outcome. Br J Surg 80 (502): 4.
Pratico C, Rizzello F, Fornarini GS, Calafiore A, Calabrese C, Campieri M, Tomassetti P, Gionchetti P (2013) Four cases of carcinoid tumour in Crohn's disease: coincidence or correlation? Int J Colorectal Dis 28 (12): 1743–1745.
Rodriguez-Bigas MA, Vasen HF, Lynch HT, Watson P, Myrhoj T, Jarvinen HJ, Mecklin JP, Macrae F St, John DJ, Bertario L, Fidalgo P, Madlensky L, Rozen P (1998) Characteristics of small bowel carcinoma in hereditary nonpolyposis colorectal carcinoma. International Collaborative Group on HNPCC. Cancer 83 (2): 240–244.
Safioleas MC, Moulakakis KG, Kontzoglou K, Stamoulis J, Nikou GC, Toubanakis C, Lygidakis NJ (2005) Carcinoid tumors of the appendix. Prognostic factors and evaluation of indications for right hemicolectomy. Hepatogastroenterology 52 (61): 123–127.
Spunt SL, Pratt CB, Rao BN, Pritchard M, Jenkins JJ, Hill DA, Cain AM, Pappo AS (2000) Childhood carcinoid tumors: the St Jude Children's Research Hospital experience. J Pediatric Surg 35 (9): 1282–1286.
Trufant J, Kurz W, Frankel A, Muthusamy V, McKinnon W, Greenblatt M, Lazar A, Cook D, Bosenberg M (2012) Familial multiple pilomatrixomas as a presentation of attenuated adenomatosis polyposis coli. J Cutan Pathol 39 (4): 440–443.
West NE, Wise PE, Herline AJ, Muldoon RL, Chopp WV, Schwartz DA (2007) Carcinoid tumors are 15 times more common in patients with Crohn's disease. Inflamm Bowel Dis 13 (9): 1129–1134.
Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, Abdalla EK, Fleming JB, Vauthey JN, Rashid A, Evans DB (2008) One hundred years after ‘carcinoid’: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 26 (18): 3063–3072.
Acknowledgements
Marjolijn Jongmans and Illja Diets are sponsored by the KiKa Foundation (project KiKa-127). Esmé Waanders is a KWF–fellow from the Dutch Cancer Society (KUN2012–5366).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Diets, I., Nagtegaal, I., Loeffen, J. et al. Childhood neuroendocrine tumours: a descriptive study revealing clues for genetic predisposition. Br J Cancer 116, 163–168 (2017). https://doi.org/10.1038/bjc.2016.408
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.408
