
Abstract
Background:
We performed a retrospective nationwide study to explore age as a prognostic factor in synovial sarcoma patients.
Methods:
Data on 613 synovial sarcoma patients were obtained from the Netherlands Cancer Registry. The prognostic relevance of age groups (children, adolescent and young adults (AYAs), adults, and elderly) was estimated by Kaplan–Meier survival curves and multivariable Cox-proportional hazards modelling.
Results:
A total of 461 patients had localised disease at diagnosis. The 5-year overall survival (OS) was 89.3±4.6%, 73.0±3.8%, 54.7±3.6%, and 43.0±7.0% in children (n=54), AYAs (n=148), adults (n=204), and elderly (n=55), respectively. Treatment modalities had no significant effect on survival in the univariable analysis. Multivariable analysis identified age at diagnosis, tumour localisation, and tumour size as significant factors affecting OS. Both tumour localisation and size were equally distributed over the age groups.
Conclusions:
We show that outcome of synovial sarcoma patients significantly decreases with age regardless of primary tumour site, size, and treatment.
Similar content being viewed by others


Main
Synovial sarcoma is a soft tissue sarcoma occurring at all ages. In the Netherlands, the standard treatment for localised disease in adults consists of radical surgery, with adjuvant radiotherapy on indication (marginal excision, R1 resection) or neo-adjuvant radiotherapy in case radical resection is deemed unattainable beforehand. Neo-adjuvant chemotherapy may be indicated when extensive down staging is necessary for the prospect of a radical resection. Adjuvant chemotherapy is mainly given within the context of a clinical trial, with a lack of convincing data substantiating survival benefit (ESMO/European SarcomaNetwork Working Group, 2014). In paediatric patients, however, patients are treated according to international (European or Children’s Oncology Group (COG)) non-rhabdomyosarcoma protocols as long as the children are candidates for such protocols and protocols are active at the time a patient presents with synovial sarcoma. The current epidemiological analysis reflects real-life management. Stage at diagnosis is a known prognostic factor, with a reported 69% 10-year cancer-specific survival in patients with local disease at diagnosis, dropping to 8.9% in patients with metastasis at diagnosis (Sultan et al, 2009). In addition to stage at diagnosis, several retrospective studies in patients with localised disease have looked at age as a prognostic factor for survival, with children having a more favourable outcome compared with adult synovial sarcoma patients. The association between age at diagnosis and survival has also been reported in a variety of other sarcomas (Ferrari et al, 2011). The present retrospective nationwide study aimed to investigate a broader range of age groups, including elderly patients, thereby explicitly exploring whether other factors such as tumour size, tumour localisation, and treatment modalities modified age-related survival differences.
Patients and methods
Data source and study population
The data were obtained from the Netherlands Cancer Registry (NCR). Cases were registered following notification by the Dutch Pathology Network (PALGA), supplemented by annual record linkage with the national hospital discharge database. Information on patient (age at diagnosis and sex) and tumour characteristics (primary tumour site, histology, size, and date of diagnosis) as well as hospital characteristics and treatment modality (type of primary treatment and resection status) was collected from hospital records by trained registrars. Follow-up information on vital status was obtained through linkage with the Municipal Personal Records Database (GBA). We obtained consent for the design, data abstraction process, as well as storage protocols from the national supervisory committee of the NCR. All patients histologically diagnosed with synovial sarcoma between 1989 and 2013 were included. The diagnosis was based on the local pathologist’s diagnosis, often complemented with histological review in regional sarcoma panels. No information on molecular markers, including the X;18 translocation, was available. For survival analyses, we only included patients diagnosed with localised disease at presentation. Patients who were not treated according to standard care (i.e., did not receive surgery as part of their primary treatment) were also excluded from the analyses, as they were considered unrepresentative. Patients were classified as children (<18 years), adolescents and young adults (AYAs; 18–34 years), adults (35–64 years), and elderly (⩾65 years), as they reflect the current Dutch situation of oncological age groups in clinical care. Treatment was divided into (1) surgery only, (2) surgery and (neo-)adjuvant radiotherapy, with or without chemotherapy, (3) surgery and (neo)adjuvant chemotherapy, and (4) no surgical treatment at all. We defined patients receiving surgery combined with chemotherapy as a separate group. We expected this to be a common treatment option in children as adjuvant chemotherapy is not a standard treatment for adult synovial sarcoma patients in the Netherlands, and could therefore be of influence on age-related survival. Due to the small patient numbers, we were limited in our ability to define treatment groups. Consequently, we grouped together who received (neo)adjuvant radiotherapy and patients receiving (neo)adjuvant radio- and chemotherapy.
Data analyses and statistics
Distribution of descriptive characteristics over the age groups was evaluated with Chi-square tests. Crude survival rates, 5- and 10-year overall survival (OS), were calculated using the Kaplan–Meier method, and the log-rank test pooled over the strata was used to compare the OS curves. In addition, relative survival (RS) analyses were performed as an approximation of disease-specific survival (Dickman et al, 2004), with OS being corrected for expected mortality according to annual life tables of the general population matched on age, gender, and calendar year (annually retrieved from Statistics Netherlands).
To identify independent prognostic factors for survival, we developed Cox proportional hazards models for which factors were selected on the basis of both clinical plausibility and significance in univariable analyses (P<0.1). In the multivariable analysis, we evaluated whether these factors modified the association between age and survival. Variables were considered confounders and included in the model on the basis of the log-likelihood test.
All statistical analyses were two-sided. Except for the selection of variables for the multivariable analysis, a P-value of <0.05 was considered significant. Statistical calculations and survival curves were generated by using IBM SPSS Statistics version 20 (Armonk, NY, USA) and Stata 13.0 (Stata Corp, College Station, TX, USA). Missing data on histological subtype, tumour size, and tumour depth were considered missing at random, and were therefore imputed under fully conditional specification, using the MI command in Stata. On the basis of variables used in the regression analysis and those predictive of missing values, we generated 50 data sets using chains of 100 iterations. Convergence of the imputations was checked graphically, and the Cox models were built using both the imputed data set and the data set restricted to cases with complete data for comparative purposes.
Results
In total, 613 synovial sarcoma patients were retrieved. At diagnosis, 461 patients (75.2%) were confirmed with localised disease (Table 1). Patients had a median age of 38.0 years (range 2–89). Surgery was performed in 416 (90.2%) of the patients, which was most often combined with radiotherapy (n=219, 47.5%). This group included patients who received both radio- and chemo-therapy (n=49). Surgery combined with chemotherapy alone was performed in a minority of the patients, although this treatment modality was provided three times more often in children (20.4%) than in the adult age groups (6.9% combined). Surgery was not undertaken in a minority of children (3.7%). Interestingly, this proportion was significantly higher in all adult patients (10.6%). Among the elderly, more than a quarter of patients did not undergo surgery (25.5%).
Compared with general hospitals, academic centres treated a significantly higher proportion of children (16.5% vs 7.3%; P=0.005). In addition, academic centres more often treated larger tumours (56.9% vs 40.9%; P=0.003) and tumours that were located deeply (54.6% vs 38.1%; P=0.031).
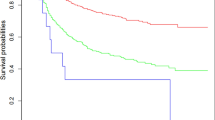
Patients’ age showed a gradual decline in OS over the years. Overall, patients had a 5- and 10-year OS rate of 63.5±2.4% and 53.8±2.6%, respectively. Subdivided in the different age groups, the 5- and 10-year OS rates were <18 years: 89.3±4.6% and 77.0±6.9%, 18–34 years: 73.0±3.8% and 64.0±4.3%, 35–64 years: 54.7±3.6% and 47.5±3.7%, and ⩾65 years: 43.0±7.0% and 28.4±7.1% (log-rank test: P<0.001) (Figure 1). RS rates did not show large discrepancies with the estimates for OS except for the elderly: 5- and 10-year RS rates were 52.0% and 46.1%, respectively.

Kaplan–Meier survival curves. The Kaplan–Meier curves of overall survival for (A) age, (B) primary tumour size, (C) primary tumour localisation, and (D) treatment.
Univariable analyses (Supplementary Table 1) identified age at diagnosis, tumour size, tumour depth, and site of origin as having a significant impact on the prognosis of synovial sarcoma patients (P<0.1), and these factors were included in the multivariable analysis. In the multivariable analysis (Supplementary Figure 1), age turned out to be an independent prognostic indicator for OS, with increasing age at diagnosis contributing to a higher risk of death. Compared with children, AYAs had a hazard ratio (HR) of 2.29 (95% CI 1.10–4.77), while this was 4.10 for adults (95%CI 2.03–8.29) and 6.18 in the elderly (95% CI 2.83–13.49). In addition, patients with a larger tumour size did significantly worse compared with tumours ⩽5cm (HR 2.89; 95% CI 1.94–4.32). Considering the primary site, patients with their primary tumour located in the trunk had the worst prognosis (HR 2.07; 95% CI 1.39–3.07). Patients with their tumour located in the head or neck (HR 1.30; 95% CI 0.73–2.34) or other sites (HR 1.75; 95%CI 0.82–3.71) had similar survival rates as patients with a tumour in the extremity. Tumour depth had no significant impact (HR 1.44; 95% CI 0.80–2.62).
Discussion
To our knowledge, this is the largest study reporting on age-related survival in synovial sarcoma patients with localised disease. Several smaller studies have attempted to demonstrate this age-related survival effect in patients with localised synovial sarcoma. Interestingly, almost none of these studies, all with different cutoff points, were able to detect a significant prognostic effect of age on patient outcome (Table 2). Nonetheless, a trend towards worse survival at higher age may be presumed, and our study demonstrated a significant survival difference across age groups in synovial sarcoma patients with localised disease at diagnosis. As no information was available on disease-specific survival, we also calculated RS rates to account for the general effect of age on mortality. The association of lower RS rates accompanying increasing age was also observed here. The drop in survival rates between 5 and 10 years across all age groups confirms the occurrence of late relapses in SS. In addition, the data show that this phenomenon occurs in all age groups.
The multivariable analysis identified older age, larger tumour size, and primary tumour localisation in the trunk as independent prognostic factors for worse OS in synovial sarcoma patients. As both tumour size and primary tumour localisation were equally distributed over the age groups, we do not believe that they accounted for the observed age-related survival effect.
Main limitations of this study concern the retrospective character of the data collection, and the long period over which data were resembled in which molecular diagnostics were first introduced and subsequently improved, which could have improved the final diagnostics. However, we do expect misdiagnosed cases to be evenly distributed across all age groups.
As this study is national-wide registry based, it is based on diagnosis made by pathologist from different hospitals, of whom many will not be sarcoma-dedicated. This could possibly explain the large proportion of pathology reports that lacked information on the synovial sarcoma subtype. The same is true with respect to residual disease status where reporting was inadequate in more than half of the surgical resections (51.9%). In addition, since the SYT-SSX translocation test has become a standard procedure only in recent years, translocations status is not yet included in the NCR database. As the first steps in sarcoma care centralisation have only been taking place in the Netherlands since 2012, improvements in pathology reporting are to be expected in the forthcoming years. Synovial sarcomas may well consist of hitherto undetermined subtypes, with different types occurring at different ages. Therefore, we hypothesise that tumour-genetic differences underlie the age effect, which has also been suggested in the study by Lagarde et al (2013), who showed increased chromosome instability in adult vs paediatric synovial sarcoma patients.
In conclusion, our study demonstrated that outcomes of patients with synovial sarcoma significantly decrease with age regardless of primary tumour site, size, and treatment. However, none of the variables included in this study seems to provide an adequate explanation for the observed difference in survival. The results of this study are of utmost importance when designing future clinical studies for localised synovial sarcoma, taking age as a prognostic factor into account. Further exploration of differences in tumour biology between different age groups may aid in adapting new treatments directed at tumour-specific characteristics.
Change history
01 December 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Al-Hussaini H, Hogg D, Blackstein ME, O'sullivan B, Catton CN, Chung PW, Griffin AM, Hodgson D, Hopyan S, Kandel R, Ferguson PC, Wunder JS, Gupta AA (2011) Clinical features, treatment, and outcome in 102 adult and pediatric patients with localized high-grade synovial sarcoma. Sarcoma 2011: 231789.
Brennan B, Stevens M, Kelsey A, Stiller CA (2010) Synovial sarcoma in childhood and adolescence: a retrospective series of 77 patients registered by the Children's Cancer and Leukaemia Group between 1991 and 2006. Pediatr Blood Cancer 55: 85–90.
Dickman PW, Sloggett A, Hills M, Hakulinen T (2004) Regression models for relative survival. Stat Med 23: 51–64.
Ferrari A, De Salvo GL, Brennan B, Van Noesel MM, De Paoli A, Casanova M, Francotte N, Kelsey A, Alaggio R, Oberlin O, Carli M, Ben-Arush M, Bergeron C, Merks JH, Jenney M, Stevens MC, Bisogno G, Orbach D (2014) Synovial sarcoma in children and adolescents: the European pediatric Soft tissue sarcoma Study Group prospective trial (EpSSG NRSTS 2005). Ann Oncol 26: 567–572.
Ferrari A, Gronchi A, Casanova M, Meazza C, Gandola L, Collini P, Lozza L, Bertulli R, Olmi P, Casali PG (2004) Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer 101: 627–634.
Ferrari A, Sultan I, Huang TT, Rodriguez-Galindo C, Shehadeh A, Meazza C, Ness KK, Casanova M, Spunt SL (2011) Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database. Pediatr Blood Cancer 57: 943–949.
ESMO/European Sarcoma Network Working Group (2014) Soft tissue and visceral sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 25 (Suppl 3): iii102–iii112.
Guadagnolo BA, Zagars GK, Ballo MT, Patel SR, Lewis VO, Pisters PW, Benjamin RS, Pollock RE (2007) Long-term outcomes for synovial sarcoma treated with conservation surgery and radiotherapy. Int J Radiat Oncol Biol Phys 69: 1173–1180.
Lagarde P, Przybyl J, Brulard C, Perot G, Pierron G, Delattre O, Sciot R, Wozniak A, Schoffski P, Terrier P, Neuville A, Coindre JM, Italiano A, Orbach D, Debiec-Rychter M, Chibon F (2013) Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J Clin Oncol 31: 608–615.
Palmerini E, Staals EL, Alberghini M, Zanella L, Ferrari C, Benassi MS, Picci P, Mercuri M, Bacci G, Ferrari S (2009) Synovial sarcoma: retrospective analysis of 250 patients treated at a single institution. Cancer 115: 2988–2998.
Sultan I, Rodriguez-Galindo C, Saab R, Yasir S, Casanova M, Ferrari A (2009) Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: an analysis of 1268 patients. Cancer 115: 3537–3547.
Tarkan Y, Erkan A, Selcuk ES, Mehmet K, Tugba KF, Ozlem US, Suleyman A, Tulay A, Bilge A, Ramazan Y, Yusuf G, Mevlude I, Umut D, Ilhan O, Abdurrahman I, Alper S, Dogan U, Necati A, Berna O, Gok DA, Ugur Y, Mahmut G (2014) Clinical and pathological features of patients with resected synovial sarcoma: a multicenter retrospective analysis of the Anatolian Society of Medical Oncology. J Cancer Res Ther 10: 73–78.
Trassard M, Le Doussal V, Hacene K, Terrier P, Ranchere D, Guillou L, Fiche M, Collin F, Vilain MO, Bertrand G, Jacquemier J, Sastre-Garau X, Bui NB, Bonichon F, Coindre JM (2001) Prognostic factors in localized primary synovial sarcoma: a multicenter study of 128 adult patients. J Clin Oncol 19: 525–534.
Yaser S, Salah S, Al-Shatti M, Abu-Sheikha A, Shehadeh A, Sultan I, Salem A, Sughayer M, Al-Loh S, Al-Mousa A (2014) Prognostic factors that govern localized synovial sarcoma: a single institution retrospective study on 51 patients. Med Oncol 31: 958.
Acknowledgements
We thank the Synovial Sarcoma Research Foundation for their financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper was presented orally at the 19th annual meeting of the Connective Tissue Oncology Society (CTOS) in Berlin, 2014.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Vlenterie, M., Ho, V., Kaal, S. et al. Age as an independent prognostic factor for survival of localised synovial sarcoma patients. Br J Cancer 113, 1602–1606 (2015). https://doi.org/10.1038/bjc.2015.375
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.375
Keywords
This article is cited by
-
Complete resection of a giant intrapericardial cardiac synovial sarcoma
Journal of Cardiothoracic Surgery (2024)
-
Establishment and characterization of NCC-SS5-C1: a novel patient-derived cell line of synovial sarcoma
Human Cell (2022)
-
Prognostic factors of the synovial sarcoma of the extremities: imaging does matter
European Radiology (2022)
-
Efficacy of perioperative chemotherapy for synovial sarcoma: a retrospective analysis of a Nationwide database in Japan
BMC Cancer (2021)
-
Magnetic resonance imaging findings of synovial sarcoma in children: location-dependent differences
Pediatric Radiology (2021)

{kind=link}