
Abstract
Therapy resistance and tumour relapse after drug therapy are commonly explained by Darwinian selection of pre-existing drug-resistant, often stem-like cancer cells resulting from random mutations. However, the ubiquitous non-genetic heterogeneity and plasticity of tumour cell phenotype raises the question: are mutations really necessary and sufficient to promote cell phenotype changes during tumour progression? Cancer therapy inevitably spares some cancer cells, even in the absence of resistant mutants. Accumulating observations suggest that the non-killed, residual tumour cells actively acquire a new phenotype simply by exploiting their developmental potential. These surviving cells are stressed by the cytotoxic treatment, and owing to phenotype plasticity, exhibit a variety of responses. Some are pushed into nearby, latent attractor states of the gene regulatory network which resemble evolutionary ancient or early developmental gene expression programs that confer stemness and resilience. By entering such stem-like, stress-response states, the surviving cells strengthen their capacity to cope with future noxious agents. Considering non-genetic cell state dynamics and the relative ease with which surviving but stressed cells can be tipped into latent attractors provides a foundation for exploring new therapeutic approaches that seek not only to kill cancer cells but also to avoid promoting resistance and relapse that are inherently linked to the attempts to kill them.
Similar content being viewed by others

Main
The failure of generic chemotherapy as well as targeted therapy, embodied by drug resistance and tumour relapse, demands a broader understanding of the tumour’s response to therapeutic perturbation, which is more complex than the binary outcome – ‘responding’ (e.g., tumour shrinks) vs ‘non-responding’. A more encompassing approach has to take into account the complexity of the regulatory networks that govern cell phenotype changes because the very same cellular machinery that affords a single metazoan genome the ability to produce the multitude of distinct, stable phenotypic cell states also accounts for the broad behavioural repertoire of cancer cells.
The current explanation for the development of therapy resistance and tumour recurrence invokes genomic alterations and selection of mutant cells carrying a relevant mutation acquired by chance before therapy (Nowell, 1976; Yachida et al, 2010; Sottoriva et al, 2013). Such somatic Darwinian selection leads to a shift in cell population composition but does not involve individual cells actively changing their phenotype: the mutant cells are passively selected by the therapy and overtake the cell population. Whole-genome sequencing of tumours with reconstruction of progression history (Yachida et al, 2010; Sottoriva et al, 2013) as well as the finding that ‘driver mutations’ affecting oncogenes within the same pathway are mutually exclusive (Sherr, 1996; DeNicola et al, 2011; Nakasone et al, 2012; Rosenzweig, 2012) offer convincing molecular footprints of Darwinian somatic evolution. The molecular nature of genetic mutations, such as point mutations that alter the target domain of protein kinases where selective inhibitor drugs bind (Vogelstein and Kinzler, 1993; DeNicola et al, 2011; Hanahan and Weinberg, 2011; Nakasone et al, 2012; Rosenzweig, 2012), also provide evidence of Darwinian selection. Accordingly, phenotype innovation during tumour progression, such as development of drug resistance or acquisition of any hallmark of cancer, is now, by default, explained by a genetic mutation (Vogelstein and Kinzler, 1993; Hanahan and Weinberg, 2011). However, a more recent study (Sottoriva et al, 2015) revealed the possibility of neutral evolution, which complicates the interpretation of genomic mutations found in the tumour tissue. In this process, which is well familiar to evolution biologists, most mutations are not selected for but instead accumulate as they are carried along and may by chance become sufficiently predominant to be detectable owing to genetic drift in relatively small cell populations.
In any case, it has long been suspected that driver mutations may only be part of the equation. The scheme of Darwinian evolution relies on the tacit assumption of a rigid one-to-one mapping from genotype to phenotype, such that a phenotype innovation can only result directly from a genomic mutation, which are random in nature but can be selected for. This line of thought ignores the diversity of cell phenotypes that can be generated by a single metazoan genome. In fact, a long line of evidence points to mechanisms that defy the scheme of genetic mutation/selection as the driver of tumour progression. The recent spate of cancer genome sequencing studies has reopened our eyes to the flexibility in the genotype–phenotype mapping, pushing us to think beyond the paradigm of mutation/selection. Such flexibility re-emerges under the term ‘phenotype plasticity’ and entails a departure from the classical mutations/selection scheme as an agent of tumour progression. The case of ependymoma without mutation (Mack et al, 2014; Shlush et al, 2014) or the reappearance of early clones, which if progression were to follow the Darwinian paradigm of selection would have been outcompeted by fitter clones (Nowak, 2006; Hoek and Goding, 2010; Roesch et al, 2010; Sharma et al, 2010; Chaffer and Weinberg, 2011; Shlush et al, 2014; Sun et al, 2014), underscore the need to embrace the non-uniqueness in the genotype–phenotype mapping and limited efficacy of selection.
In this review, we consider non-genetic cell phenotype plasticity as a central process in therapy resistance. We take into account the ability of cells to produce discretely distinct phenotypes, switch between them without genomic alterations and inherit the new phenotype non-genetically across cell generations (Brock et al, 2009). Here, we explain the underlying biology of cell state dynamics and the formal framework of cancer attractors that link cell phenotype dynamics to first principles of dynamical systems theory and offer a conceptual aid for comprehending mutation-independent tumour progression. As illustration, we present examples of therapy-induced drug resistance and discuss implications for new therapies.
Attractors and non-genetic switching on the epigenetic landscape
In the past years, cell fate-switching between a more stem-like, therapy-resistant state and a more differentiated, drug-sensitive state has been observed in a number of tumour cell populations (Hoek and Goding, 2010; Sharma et al, 2010; Chaffer and Weinberg, 2011; DeNicola et al, 2011; Nakasone et al, 2012; Sun et al, 2014). The reversibility of such switching and the clonality of the populations in which both states coexist indicate that this cell phenotype change is not caused by mutations.
Because of their longevity in the tissue, normal stem cells are endowed with increased xenobiotic resistance, in part, due to the expression of efflux pumps (Zhou et al, 2001; Challen and Little, 2006; Kenyon and Gerson, 2007; DeNicola et al, 2011; Nakasone et al, 2012; Rosenzweig, 2012), more efficient DNA repair (Kenyon and Gerson, 2007) and they also orchestrate tissue changes during wound healing. Thus, the stem-like state is deeply linked to resilience and stress response – a relationship that appears to hold for their neoplastic counterpart, the cancer stem cells (CSC) (Dean et al, 2005; Donnenberg and Donnenberg, 2005; Medema, 2013). A fundamental biological fact is then that the axis of cell phenotype with respect to drug tolerance is aligned with that of cellular stemness.
Here, we will rely on the example of a two-state dichotomy that can often be observed by measuring expression status of a single protein marker X (e.g., XHigh and XLow). This may appear simplistic, but serves well for the conceptualisation of cell state dynamics (Figure 1). Phenotype switching between the cancer stem-cell state, here XHigh (where X can be one of the stem-cell markers, such as CD117, MDR1, CD44, ALDH1; Hanahan and Weinberg, 2011; Medema, 2013), and the more differentiated, here XLow, state epitomises non-genetic plasticity of cancer cells. The underlying principles are the same as those that govern cell phenotype changes in development and tissue homeostasis: the acquisition of a stable and distinct cell phenotype governed by a coordinated gene expression pattern that can be passed along to daughter cells upon division. Although trivial, one often forgets that multiple distinct phenotypic states can be produced by the very same genome, which is the foundation for non-genetic switching between functional states during tumour progression.

State transition and non-genetic plasticity. The cellular transition from the state with XHigh (red) to XLow (blue) and vice versa can be thought of as a state transition between the two subattractors on the epigenetic landscape (see Box 1). The reversibility of such switching and the clonality of the populations in which both states coexist indicate that such cell phenotype changes are not caused by mutations. As in multistable systems, the transition is noise driven but modulated by external conditions, including the presence of the drug. Note that by monitoring one dimension of the gene expression state space, for example, X=MDR1 expression, we are able to observe cell transition only as a projection (horizontal axis) and do not know what happens in orthogonal (non-observable) dimensions.
The existence of multiple, almost discretely distinct, stable states, each characterised by its own stable gene expression profiles that is produced by the very same genome, can be explained by the concept of multistability. Herein, the stable states are the so-called attractor states – the potential wells in an energy landscape (Figure 1 and Box 1).
With this formal conceptual framework, non-genetic switching between phenotypic states can be represented as switching between distinct attractor states – a conceptualisation well familiar to physical scientists. The key properties that distinguish such non-genetic state switching from a mutation-driven phenotype alteration are that the former (i) occurs much more frequently than mutations, (ii) is in principle reversible and (iii) is linked to regulatory dynamics of the GRN, and thus has features of non-randomness. The last property is a central distinctive feature of non-genetic phenotype transitions: unlike undirected (blinded) mutations of DNA, non-genetic switching between attractor states is likely to turn on existing (evolved) cellular programs and to be influenced by external cues. This framework could explain why tumour cells can so readily acquire complex developmental programs, such as a stem-like state or any of the ‘hallmarks of cancer’ (Hoek and Goding, 2010; Hanahan and Weinberg, 2011). Their gradual Darwinian evolution de novo is not required because these states have equivalents in normal development, homeostasis and regeneration.
For such mutation-less cell phenotype switching in tumour progression, we can distinguish between two scenarios: (1) spontaneous (random) phenotype switching between a stem-like, resistant and a more differentiated (less aggressive) state and (2) externally induced (directed) switching to the stem-like state. The switching between the more mature and the stem-like states has been intensely studied in melanoma (Sharma et al, 2010; Hoek and Goding, 2010), where the bidirectional switching, that is, the reversibility of differentiation of CSC into multiple cell types, long thought to be an one-way process, supports the emerging notion that CSCs do not represent a static mutant clone but rather a functional state that can be induced in some cells under some conditions (Gupta et al, 2009a; Dirks, 2010; Gerlinger et al, 2012).
Spontaneous state switching
The first scenario, spontaneous switching between attractors, is evident in cell population measurements of a marker X in (clonal) tumour cell cultures by flow cytometry. Such measurements can reveal the coexistence of the two states, occupied by two fractions of a clonal population, through the appearance of a bimodal distribution of the flow cytometry histogram (two ‘peaks’) (Figure 1). The presence of distinct peaks suggests (but does not prove) multistability. Such non-genetic heterogeneity must be distinguished from genetic heterogeneity caused by genomic instability (Gerlinger et al, 2012; Pisco et al, 2013).
Using fluorescent-activated cell sorting to isolate cells from one peak followed by reculturing often – but not always – shows that one sub-population (e.g., XHigh) can repopulate the other (e.g., XLow) (Chang et al, 2006; Sharma et al, 2010; Pisco et al, 2013), demonstrating spontaneous, non-genetic transitions across a sufficiently low ‘energy’ barrier. Therefore, individual cells switch in a stochastic manner from one (attractor) state (XHigh) to the other (XLow), revealing characteristic rates for transitions in either direction. The driving force and source of randomness for such state switching is gene expression noise (=fluctuations of protein levels due to the random nature of molecular reactions within small volumes in the cell) (Kaern et al, 2005; Raj and van Oudenaarden, 2008; Zhou et al, 2014). Transitions between the XHigh and XLow state, although bidirectional, are often asymmetric – reflecting the relative propensity of a given cancer cell type to differentiate into a more mature state, or of the latter to dedifferentiate back into the stem-like state. ‘Dedifferentiation’ results in the continuous production of CSCs by the more differentiated cells and has been observed in various cancers posing a challenge to therapy aimed at targeting CSC (Brock et al, 2009; Gupta et al, 2009b).
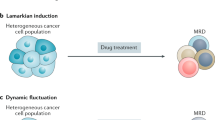
Cell population dynamics with spontaneous conversions between resistant and sensitive states can be subjected to Darwinian selection in the same manner as phenotype variability caused by mutations (Figure 2A): the stochastic but non-genetic (mutation-independent) transitions between attractor states act in the same way as mutations in Darwinian evolution in producing a phenotype variability as substrate for selection – albeit with much higher frequency and repeatedly generating a particular complex phenotype (as it is a latently ‘preprogrammed’ attractor). Thus, in the presence of cytotoxic drugs the cells in the more stem-like, more drug-tolerant state in a population will have a proliferation advantage and can be selected for if they endure over several cell generations in that stem-like state (Brock et al, 2009). Such ‘non-genetic selection’ can provide temporary resistance and allow the sub-population of these cells to expand. As these non-genetically fitter variants are present in the multimodal population due to a dynamic equilibrium, they exist by default and at much higher frequency than genetic mutants (accounting for 1% or more in a clonal population). Thus, they can jump start classical (mutation-based) somatic evolution because they guarantee that there is always a small population of cells that independently of mutations can survive the treatment. As these cells expand, they can accumulate genetic mutations that are then selected for. This scheme of preselection due to non-genetic variability followed by later genetic fixation is in essence Waddington’s genetic assimilation (Waddington, 1942; Zhou et al, 2001; Kim et al, 2002). An interesting twist that may further accelerate this process of somatic evolution is that since many anticancer drugs are genotoxic, mutation rate is further increased among these initial stem-like survivors.

Two schemes for the evolution of drug-resistant state. (A) In the first scenario, according to the Darwinian selection, the drug-resistant phenotype results from selection of resistant clones, which were produced by genetic mutations. The genetic mutation alters the genome, causing a rewiring of the gene regulatory network (GRN), which in turn changes the epigenetic landscape, creating a new phenotype (attractor state, S(XHigh)). (B) In the second scenario, the drug causes an attractor transition from the sensitive (blue) to the resistant (red) state in a multistable system (landscape with two potential wells). The GRN and the landscape remain unchanged.
Therapy-induced state switching: therapy as doubled-edged sword
In the second scenario, an external stimulation that affects signalling pathways can change the gene expression state, thus triggering a state transition between attractor states in a multistable system. In normal tissue development, attractor transitions correspond to cell differentiation and are induced by tightly regulated developmental signals that trigger transitions to lower attractor states. Of particular interest for tumour progression is the stress inflicted onto the (non-killed) cancer cells by treatment and the ensuing state transition. Given the commonalities between stress-response and stemness programmes (Zhou et al, 2001; Kim et al, 2002; Blanpain et al, 2011) and the inherent immaturity of cancer cells explained above, it is possible that cell stress imparted by cytotoxic agents actively induces a state transition specifically into a stem-like state, which actually is a stress-response state.
Such cell response is not unexpected if we consider the theoretical epigenetic landscape (Figure 2B). In this framework, there are many possible (unused) cell states in the neighbourhood of a cancer attractor, which due to the tight relationship between ontogenesis and phylogenesis likely represent evolutionary ancient, stem-like immature states. Therefore, entry into these unoccupied attractors does not require very specific signals but can be triggered by many unrelated, nonspecific cell perturbations. As normal stem cells are specialised to respond to tissue damage by providing a protective response (Blanpain et al, 2011; Huang, 2013), these nearby abnormal immature states occupied by cancer states may express similar properties. Thus, a cytotoxic, non-lethal cell stress applied to an already pathologically immature cell in a cancer attractor has a high probability to push the cell further into more immature (‘dedifferentiated’) regions of the epigenetic landscape (Huang, 2013).
A therapy-induced, directed somatic evolution towards an advantageous phenotype would correspond to a Lamarckian scheme: a new, better ‘adapted’, inheritable state is induced by an environmental input (Mayr, 1972). In view of the conceptual framework of the epigenetic landscape, this event would not be uncommon, but in contrary almost a default reaction of cancer cells to massive, near-lethal perturbations. In fact, the list of cases in which chemotherapy appears to induce either a stem-like or/and drug-resistant state is quite old and growing (Bates et al, 1989; Chin et al, 1990; Ciocca et al, 1992; Chaudhary and Roninson, 1993; Abolhoda et al, 1999; Stein, 2001; Iyer and Lehnert, 2002; Notarbartolo et al, 2002; Bortul et al, 2003; Gréen et al, 2003; Baker et al, 2005; Camphausen et al, 2005; Andarawewa et al, 2007; Xia et al, 2008; Li et al, 2009; Pajic et al, 2009; Taube et al, 2010; Lee et al, 2010; Balachandran et al, 2011; Chang et al, 2011; Ghisolfi et al, 2012; Lagadec et al, 2012; Razandi et al, 2012; Abubaker et al, 2013; Pisco et al, 2013, 2014).
The induction of multidrug resistance (MDR) proteins, the ABC family membrane pumps that mediate the active efflux of drugs and also are markers of stem cells (‘dye exclusion’), is the best documented example of an induced adaptive trait (Bates et al, 1989; Chin et al, 1990; Chaudhary and Roninson, 1993; Abolhoda et al, 1999; Yamada et al, 2000; Stein, 2001; Notarbartolo et al, 2002; Gréen et al, 2003; Baker et al, 2005; Pajic et al, 2009; Kim et al, 2012; Stein et al, 2012; Pisco et al, 2013, 2014). Wnt signalling has a central role in induced stemness and MDR upregulation in various cancer cells (Yamada et al, 2000; Lee et al, 2010; Kim et al, 2012; Stein et al, 2012; Pisco et al, 2013). In irradiated leukaemia cells, resistance to radiotherapy was acquired without ‘clonal predisposition’ (Lee et al, 2010; Lagadec et al, 2012). Similarly, ionising radiation reprogrammed differentiated breast cancer cells into CSCs via Notch (Lagadec et al, 2012) and the CSC fraction increased following cisplatin therapy in head-and-neck cancer (Nör et al, 2014). Several reports suggest chemotherapy-induced epithelial–mesenchymal transition, which has been linked to transition into a stem-like state (Dallas et al, 2009; Polyak and Weinberg, 2009; Singh and Settleman, 2010), including in colon (Dallas et al, 2009; Xue et al, 2011), gastric (Pirozzi et al, 2011; Xue et al, 2011), liver cancer (Gupta et al, 2011; Pirozzi et al, 2011) and breast cancer (Donaldson et al, 1978; Wallner and Li, 1986; Gupta et al, 2011).
Treatment-induced stress response might indeed protect the cancer cell (Iliopoulos et al, 2011; Jackson et al, 2013; Halliday et al, 2014). This protective stress response was explicitly observed by exposing cancer cells to nonlethal heat-shock before chemotherapy and was associated with the upregulation of heat-shock proteins and ABC transporters (Ciocca et al, 1992).
Most studies do not distinguish between Darwinian and Lamarckian scheme in resistance development
Often in the literature terms such as ‘acquisition’, ‘enrichment’ or ‘induction’ of stemness and resistance (Pardoll, 2012; Heams et al, 2014; Sun et al, 2014), as well as expressions such as ‘activation of alternative pathways’ that circumvent the blockade by target-selective drug or ‘adaptive resistance’(Abolhoda et al, 1999; Huang et al, 2012; Pardoll, 2012; Tenbaum et al, 2012; Das Thakur et al, 2013; Sun et al, 2014), are used in a hand-waving manner without specifying the class of mechanisms behind the development of therapy resistance. Three types of scenarios are possible: tumour progression can be driven by (i) selection of genetic mutants (the classical Darwinian ‘adaptation’); (ii) selection of cells in non-genetic, pre-existing states occupied by chance by a sub-population of cells (see Section ‘Spontaneous state switching’); (iii) a true induction of cell state transition by the therapeutic agent in individual surviving cells (see Section ‘Therapy-induced state switching: therapy as doubled-edged sword’). The former two scenarios are cell population-level processes, involving selection of inherently fitter cells. Older analyses, based on the upregulation of resistance/stemness genes at the whole-cell population level (e.g., western blot) and lacking detailed kinetics, obscure the difference between population (selection) and cell-individual (induction) phenomena (Abolhoda et al, 1999; Huang et al, 2012; Tenbaum et al, 2012; Das Thakur et al, 2013; Sun et al, 2014).
The short timing for the appearance of stem-like or MDR+ cells after treatment (hours to few days) and the regularity with which any anticancer treatment triggers a complex stress-response phenotype suggests that the non-genetic process is often behind the observed change (scenario ii or iii). However, the question whether this apparent ‘enrichment’ of stem-like cells is due to selection of cells that occupied the stem-like state or induction (conversion of an individual cell by treatment) is usually not explicitly raised. Experimental demonstration of the latter requires longitudinal monitoring of phenotype change in individual cells (Nowell, 1976; Pisco et al, 2013). The essential difference between a Lamarckian, non-genetic, induced cell response and a classical Darwinian selection (of mutants or of attractor states) is that the induced response is mounted by surviving cells that were not a priori more resistant. This conceptual difference has therapeutic consequences for preventing resistance.
Outlook: therapeutic implications beyond killing
The mechanisms of resistance described here rest on the fact that non-mutant, non-resistant, treatment-naive cancer cells can survive treatment. However, why do they survive in the first place? First, tumour therapy has to balance between killing tumour cells and avoiding host toxicity, thus limiting the dose used. Second, penetration of tumour tissue by drug molecules is not uniform, creating ample opportunity for many tumour cells survive even if they are not a priori resistant. And third, even within a clonal population, the cell-intrinsic sensitivity to drug fluctuates, as do most cellular traits, resulting in a broad statistical distribution (heterogeneity) of drug sensitivity at any time point (Huang, 2009). Therefore, therapy will almost inevitably not kill all tumour cells, independently of pre-existing genetic mutants and even when considering cell-cycle phase. However, the surviving cancer cells are not innocent bystanders: having encountered a sublethal perturbation, which is a massive blow, some of them will be pushed into nearby (unused) attractors, the dead-end sidevalleys of the epigenetic landscape, which likely, as discussed above, encode stem-like, stress-response phenotypes. This explains why non-killed but near-lethally stressed cells are not necessarily weaker but instead might become more resilient (McDunn, 2005; Huang, 2014). Hence, rather than describing tumour progression with the principle of Darwin’s ‘Survival of the fittest’, it may be more suited to think of Nietzsche’s concept that ‘What does not kill me strengthens me’.
Although progression is an intrinsic trait of malignant tumours and debulking of the primary tumour is clinically necessary to alleviate disease burden and lessen the probability of metastatic dissemination, we also need to consider the progression-stimulating effect of therapy in the non-killed cells. Therefore, tumouricidal treatment is inherently a double-edged sword. The question is: how can we maximise the desired effects of therapy while minimising the strengthening of the (unavoidable) surviving cells? We can consider two types of approaches (Figure 3): the proactive solution would be to combine cytotoxic therapy with drugs that block the Lamarckian induction of a stress response. For instance, Wnt signalling has emerged as a central pathway in stress-induced stemness and resistance in tumour and stromal cells (Corrêa et al, 2012; He et al, 2014). Inhibitors of the Wnt-β–catenin axis, currently tested as differentiating agents (Liu et al, 2013), may be used before chemo- or targeted therapy to prevent stress response in surviving cells. The second approach is more passive and seeks ‘gentle containment’: avoid triggering the stress response associated with aggressive attempts to kill. This can be achieved by a departure from the traditional maxim of ‘maximal-tolerated dose’ through a variety of scheduling (Liao et al, 2012), such as metronomic therapy (lower dose, more frequent) (Scharovsky et al, 2009) or drug holidays to allow for reversion to the sensitive state (Sun et al, 2014). Non-cytotoxic differentiation therapy (Sell, 2004) may also belong to this category.

Different anticancer therapy schemes can minimise the strengthening of surviving cells. If rather than treating the tumour using conventional, aggressive therapy that may stimulate the stressed, stem-like (=more aggressive) state, cancer is treated using alternative, gentler modification of cell growth we might be able to control the ratio between the stressed cells and the naive, non-stressed cancer cells. The idea of treating cancer as a chronic disease is to prevent the cells from transiting into the aggressive state in response to cytotoxic stress. This can be achieved by blocking transition into that state (right blue arrow) or by containing cells without cytotoxity, for example, by promoting differentiation (green arrow).
The immense non-genetic phenotypic heterogeneity of tumour cells, which entails, by statistical necessity, incompleteness of any tumouricidal treatment, and the phenotype plasticity, which allows perturbed surviving cells to aberrantly access hidden, pre-existing, pathological stem-like states, offers a new set of principles for future exploration of therapeutic options that will reach beyond the current emphasis on killing.
References
Abolhoda A, Wilson AE, Ross H, Danenberg PV, Burt M, Scotto KW (1999) Rapid activation of MDR1 gene expression in human metastatic sarcoma after in vivo exposure to doxorubicin. Clin Cancer Res 5: 3352–3356.
Abubaker K, Latifi A, Luwor R, Nazaretian S, Zhu H, Quinn MA, Thompson EW, Findlay JK, Ahmed N (2013) Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol Cancer 12: 24.
Andarawewa KL, Erickson AC, Chou WS, Costes SV, Gascard P, Mott JD, Bissell MJ, Barcellos-Hoff MH (2007) Ionizing radiation predisposes nonmalignant human mammary epithelial cells to undergo transforming growth factor induced epithelial to mesenchymal transition. Cancer Res 67: 8662–8670.
Baker EK, Johnstone RW, Zalcberg JR, El-Osta A (2005) Epigenetic changes to the MDR1 locus in response to chemotherapeutic drugs. Oncogene 24: 8061–8075.
Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, Sorenson EC, Popow R, Ariyan C, Rossi F, Besmer P, Guo T, Antonescu CR, Taguchi T, Yuan J, Wolchok JD, Allison JP, DeMatteo RP (2011) Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med 17: 1094–1100.
Bates SE, Mickley LA, Chen YN, Richert N, Rudick J, Biedler JL, Fojo AT (1989) Expression of a drug resistance gene in human neuroblastoma cell lines: modulation by retinoic acid-induced differentiation. Mol Cell Biol 9: 4337–4344.
Blanpain C, Mohrin M, Sotiropoulou PA, Passegué E (2011) DNA-damage response in tissue-specific and cancer stem cells. Stem Cell 8: 16–29.
Bortul R, Tazzari PL, Cappellini A, Tabellini G, Billi AM, Bareggi R, Manzoli L, Cocco L, Martelli AM (2003) Constitutively active Akt1 protects HL60 leukemia cells from TRAIL-induced apoptosis through a mechanism involving NF-κB activation and cFLIPL up-regulation. Leukemia 17: 379–389.
Brock A, Chang H, Huang S (2009) Non-genetic heterogeneity – a mutation-independent driving force for the somatic evolution of tumours. Nat Rev Genet 10: 336–342.
Camphausen K, Purow B, Sproull M, Scott T, Ozawa T, Deen DF, Tofilon PJ (2005) Influence of in vivo growth on human glioma cell line gene expression: convergent profiles under orthotopic conditions. Proc Natl Acad Sci USA 102: 8287–8292.
Chaffer CL, Weinberg RA (2011) A perspective on cancer cell metastasis. Science 331: 1559–1564.
Challen GA, Little MH (2006) A side order of stem cells: the SP phenotype. Stem Cells 24: 3–12.
Chang HH, Oh PY, Ingber DE, Huang S (2006) Multistable and multistep dynamics in neutrophil differentiation. BMC Cell Biol 7: 11.
Chang Q, Jurisica I, Do T, Hedley DW (2011) Hypoxia predicts aggressive growth and spontaneous metastasis formation from orthotopically grown primary xenografts of human pancreatic cancer. Cancer Res 71: 3110–3120.
Chaudhary PM, Roninson IB (1993) Induction of multidrug resistance in human cells by transient exposure to different chemotherapeutic drugs. J Natl Cancer Inst 85: 632–639.
Chin KV, Tanaka S, Darlington G, Pastan I, Gottesman MM (1990) Heat shock and arsenite increase expression of the multidrug resistance (MDR1) gene in human renal carcinoma cells. J Biol Chem 265: 221–226.
Ciocca DR, Fuqua SAW, Lock-Lim S, Toft DO, Welch WJ, McGuire WL (1992) Response of human breast cancer cells to heat shock and chemotherapeutic drugs. Cancer Res 52: 3648–3654.
Corrêa S, Binato R, Rocher Du B, Castelo-Branco MTL, Pizzatti L, Abdelhay E (2012) Wnt/β-catenin pathway regulates ABCB1 transcription in chronic myeloid leukemia. BMC Cancer 12: 303.
Dallas NA, Xia L, Fan F, Gray MJ, Gaur P, van Buren G2, Samuel S, Kim MP, Lim SJ, Ellis LM (2009) Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res 69: 1951–1957.
Dean M, Fojo T, Bates S (2005) Tumour stem cells and drug resistance. Nat Rev Cancer 5: 275–284.
DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA, Tuveson DA (2011) Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475: 106–109.
Dirks P (2010) Cancer stem cells: Invitation to a second round. Nature 466: 40–41.
Donaldson SS, Gordon LF, Hahn GM (1978) Protective effect of hyperthermia against the cytotoxicity of actinomycin D on Chinese hamster cells. Cancer Treat Rep 62: 1489–1495.
Donnenberg VS, Donnenberg AD (2005) Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J Clin Pharmacol 45: 872–877.
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366: 883–892.
Ghisolfi L, Keates AC, Hu X, Lee D-K, Li CJ (2012) Ionizing radiation induces stemness in cancer cells. PLoS One 7: e43628.
Gréen H, Lotfi K, Zackrisson A-L, Peterson C (2003) Spontaneous reversal of p-glycoprotein expression in multidrug resistant cell lines. Pharmacol Toxicol 93: 297–304.
Gupta PB, Chaffer CL, Weinberg RA (2009a) Cancer stem cells: mirage or reality? Nat Med 15: 1010–1012.
Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES (2011) Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 146: 633–644.
Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES (2009b) Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 138: 645–659.
Halliday J, Helmy K, Pattwell SS, Pitter KL, LaPlant Q, Ozawa T, Holland EC (2014) In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural–mesenchymal shift. Proc Natl Acad Sci USA 111: 5248–5253.
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674.
He K, Xu T, Xu Y, Ring A, Kahn M, Goldkorn A (2014) Cancer cells acquire a drug resistant, highly tumorigenic, cancer stem-like phenotype through modulation of the PI3K/Akt/β-catenin/CBP pathway. Int J Cancer 134: 43–54.
Heams T, Huneman P, Lecointre G, Silberstein M (2014) Handbook of Evolutionary Thinking in the Sciences. Springer: Berlin, Germany.
Hoek KS, Goding CR (2010) Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res 23: 746–759.
Huang S (2009) Non-genetic heterogeneity of cells in development: more than just noise. Development 136: 3853–3862.
Huang S (2011) The molecular and mathematical basis of Waddington’s epigenetic landscape: a framework for post-Darwinian biology? BioEssays 34: 149–157.
Huang S (2013) Genetic and non-genetic instability in tumor progression: link between the fitness landscape and the epigenetic landscape of cancer cells. Cancer Metast Rev 32: 423–448.
Huang S (2014) The war on cancer: lessons from the war on terror. Front Oncol 4: 293.
Huang S, Ernberg I, Kauffman S (2009) Cancer attractors: a systems view of tumors from a gene network dynamics and developmental perspective. Semin Cell Dev Biol 20: 869–876.
Huang S, Hölzel M, Knijnenburg T, Schlicker A, Roepman P, McDermott U, Garnett M, Grernrum W, Sun C, Prahallad A, Groenendijk FH, Mittempergher L, Nijkamp W, Neefjes J, Salazar R, Dijke Ten P, Uramoto H, Tanaka F, Beijersbergen RL, Wessels LFA, Bernards R (2012) MED12 controls the response to multiple cancer drugs through regulation of TGF-β receptor signaling. Cell 151: 937–950.
Iliopoulos D, Hirsch HA, Wang G, Struhl K (2011) Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci USA 108: 1397–1402.
Iyer R, Lehnert BE (2002) Low dose, low-LET ionizing radiation-induced radioadaptation and associated early responses in unirradiated cells. Mutat Res 503: 1–9.
Jackson TR, Salmina K, Huna A, Inashkina I, Jankevics E, Riekstina U, Kalnina Z, Ivanov A, Townsend PA, Cragg MS, Erenpreisa J (2013) DNA damage causes TP53-dependent coupling of self-renewal and senescence pathways in embryonal carcinoma cells. Cell Cycle 12: 430–441.
Kaern M, Elston TC, Blake WJ, Collins JJ (2005) Stochasticity in gene expression: from theories to phenotypes. Nat Rev Genet 6: 451–464.
Kauffman S (1971) Differentiation of malignant to benign cells. J Theoret Biol 31: 429–451.
Kenyon J, Gerson SL (2007) The role of DNA damage repair in aging of adult stem cells. Nucleic Acids Res 35: 7557–7565.
Kim M, Turnquist H, Jackson J, Sgagias M, Yan Y, Gong M, Dean M, Sharp JG, Cowan K (2002) The multidrug resistance transporter ABCG2 (breast cancer resistance protein 1) effluxes Hoechst 33342 and is overexpressed in hematopoietic stem cells. Clin Cancer Res 8: 22–28.
Kim Y, Kim KH, Lee J, Lee Y-A, Kim M, Lee SJ, Park K, Yang H, Jin J, Joo KM, Lee J, Nam D-H (2012) Wnt activation is implicated in glioblastoma radioresistance. Lab Invest 92: 466–473.
Lagadec C, Vlashi E, Donna Della L, Dekmezian C, Pajonk F (2012) Radiation-induced reprogramming of breast cancer cells. Stem Cells 30: 833–844.
Lee G-Y, Shim J-S, Cho B, Jung J-Y, Lee D-S, Oh I-H (2010) Stochastic acquisition of a stem cell-like state and drug tolerance in leukemia cells stressed by radiation. Int J Hematol 93: 27–35.
Li Q-Q, Xu J-D, Wang W-J, Cao X-X, Chen Q, Tang F, Chen Z-Q, Liu X-P, Xu Z-D (2009) Twist1-mediated adriamycin-induced epithelial–mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin Cancer Res 15: 2657–2665.
Liao D, Estévez-Salmeron L, Tlsty TD (2012) Conceptualizing a tool to optimize therapy based on dynamic heterogeneity. Phys Biol 9: 065005.
Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, Kasibhatla S, Schuller AG, Li AG, Cheng D, Li J, Tompkins C, Pferdekamper A, Steffy A, Cheng J, Kowal C, Phung V, Guo G, Wang Y, Graham MP, Flynn S, Brenner JC, Li C, Villarroel MC, Schultz PG, Wu X, McNamara P, Sellers WR, Petruzzelli L, Boral AL, Seidel HM, McLaughlin ME, Che J, Carey TE, Vanasse G, Harris JL (2013) Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci USA 110: 20224–20229.
Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stütz AM, Wang X, Gallo M, Garzia L, Zayne K, Zhang X, Ramaswamy V, Jäger N, Jones DTW, Sill M, Pugh TJ, Ryzhova M, Wani KM, Shih DJH, Head R, Remke M, Bailey SD, Zichner T, Faria CC, Barszczyk M, Stark S, Seker-Cin H, Hutter S, Johann P, Bender S, Hovestadt V, Tzaridis T, Dubuc AM, Northcott PA, Peacock J, Bertrand KC, Agnihotri S, Cavalli FMG, Clarke I, Nethery-Brokx K, Creasy CL, Verma SK, Koster J, Wu X, Yao Y, Milde T, Sin-Chan P, Zuccaro J, Lau L, Pereira S, Castelo-Branco P, Hirst M, Marra MA, Roberts SS, Fults D, Massimi L, Cho YJ, Van Meter T, Grajkowska W, Lach B, Kulozik AE, Deimling von A, Witt O, Scherer SW, Fan X, Muraszko KM, Kool M, Pomeroy SL, Gupta N, Phillips J, Huang A, Tabori U, Hawkins C, Malkin D, Kongkham PN, Weiss WA, Jabado N, Rutka JT, Bouffet E, Korbel JO, Lupien M, Aldape KD, Bader GD, Eils R, Lichter P, Dirks PB, Pfister SM, Korshunov A, Taylor MD (2014) Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506: 445–450.
Mayr E (1972) Lamarck revisited. J Hist Biol 5: 55–94.
McDunn JE (2005) That which does not kill you makes you stronger: a molecular mechanism for preconditioning. Sci STKE 2005: pe34–pe34.
Medema JP (2013) Cancer stem cells: the challenges ahead. Nat Cell Biol 15: 338–344.
Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, Fein M, Rasch MG, Tan YX, Qiu J, Park J, Sinha P, Bissell MJ, Frengen E, Werb Z, Egeblad M (2012) Imaging tumor–stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell 21: 488–503.
Notarbartolo M, Cervello M, Dusonchet L, Cusimano A, D'Alessandro N (2002) Resistance to diverse apoptotic triggers in multidrug resistant HL60 cells and its possible relationship to the expression of P-glycoprotein, Fas and of the novel anti-apoptosis factors IAP (inhibitory of apoptosis proteins). Cancer Lett 180: 91–101.
Nowak MA (2006) Evolutionary Dynamics. Harvard University Press: Cambridge, MA, USA.
Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194: 23–28.
Nör C, Zhang Z, Warner KA, Bernardi L, Visioli F, Helman JI, Roesler R, Nör JE (2014) Cisplatin induces Bmi-1 and enhances the stem cell fraction in head and neck cancer. Neoplasia 16: 137–146.
Pajic M, Iyer JK, Kersbergen A, van der Burg E, Nygren AOH, Jonkers J, Borst P, Rottenberg S (2009) Moderate increase in Mdr1a/1b expression causes in vivo resistance to doxorubicin in a mouse model for hereditary breast cancer. Cancer Res 69: 6396–6404.
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12: 252–264.
Pirozzi G, Tirino V, Camerlingo R, Franco R (2011) Epithelial to mesenchymal transition by TGFβ-1 induction increases stemness characteristics in primary non small cell lung cancer cell line. PLoS One 6: e21548.
Pisco AO, Brock A, Zhou J, Moor A, Mojtahedi M, Jackson D, Huang S (2013) Non-Darwinian dynamics in therapy-induced cancer drug resistance. Nat Commun 4: 2467.
Pisco AO, Jackson DA, Huang S (2014) Reduced intracellular drug accumulation in drug-resistant leukemia cells is not only solely due to MDR-mediated efflux but also to decreased uptake. Front Oncol 4: 306.
Polyak K, Weinberg RA (2009) Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 9: 265–273.
Raj A, van Oudenaarden A (2008) Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 135: 216–226.
Razandi M, Pedram A, Jordan VC, Fuqua S, Levin ER (2012) Tamoxifen regulates cell fate through mitochondrial estrogen receptor beta in breast cancer. Oncogene 32: 3274–3285.
Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T, Herlyn M (2010) A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 141: 583–594.
Rosenzweig SA (2012) Acquired resistance to drugs targeting receptor tyrosine kinases. Biochem Pharmacol 83: 1041–1048.
Scharovsky OG, Mainetti LE, Rozados VR (2009) Metronomic chemotherapy: changing the paradigm that more is better. Curr Oncol 16: 7–15.
Sell S (2004) Stem cell origin of cancer and differentiation therapy. Crit Rev Oncol Hematol 51 (1): 1–28.
Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, Wong K-K, Brandstetter K, Wittner B, Ramaswamy S, Classon M, Settleman J (2010) A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141: 69–80.
Sherr CJ (1996) Cancer cell cycles. Science 274: 1672–1677.
Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW, McLeod JL, Doedens M, Medeiros JJF, Marke R, Kim HJ, Lee K, McPherson JD, Hudson TJ HALT Pan-Leukemia Gene Panel Consortium Brown AMK, Yousif F, Trinh QM, Stein LD, Minden MD, Wang JCY, Dick JE (2014) Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 506: 328–333.
Singh A, Settleman J (2010) EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29: 4741–4751.
Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J, Marjoram P, Siegmund K, Press MF, Shibata D, Curtis C (2015) A Big Bang model of human colorectal tumor growth. Nat Genet 47: 209–216.
Sottoriva A, Spiteri I, Piccirillo SGM, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavaré S (2013) Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA 110: 4009–4014.
Stein U (2001) Hyperthermia-induced nuclear translocation of transcription factor YB-1 leads to enhanced expression of multidrug resistance-related abc transporters. J Biol Chem 276: 28562–28569.
Stein U, Fleuter C, Siegel F, Smith J, Kopacek A, Scudiero DA, Hite KM, Schlag PM, Shoemaker RH, Walther W (2012) Impact of mutant β-catenin on ABCB1 expression and therapy response in colon cancer cells. Br J Cancer 106: 1395–1405.
Sun C, Wang L, Huang S, Heynen GJJE, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM, Zecchin D, Hobor S, Bajpe PK, Lieftink C, Mateus C, Vagner S, Grernrum W, Hofland I, Schlicker A, Wessels LFA, Beijersbergen RL, Bardelli A, Di Nicolantonio F, Eggermont AMM, Bernards R (2014) Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 508: 118–122.
Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, Hollier BG, Ram PT, Lander ES, Rosen JM, Weinberg RA, Mani SA (2010) Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci USA 107: 15449–15454.
Tenbaum SP, Ordonez-Moran P, Puig I, Chicote I, Arques O, Landolfi S, Fernandez Y, Herance JR, Gispert JD, Mendizabal L, Aguilar S, Cajal SR, Schwartz SJ, Vivancos A, Espin E, Rojas S, Baselga J, Tabernero J, Munoz A, Palmer HG (2012) Beta-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat Med 18: 892–901.
Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, Stuart DD (2013) Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 494: 251–255.
Vogelstein B, Kinzler KW (1993) The multistep nature of cancer. Trends Genet 9: 138–141.
Waddington CH (1942) Canalization of development and the inheritance of acquired characters. Nature 3811: 563–565.
Waddington CH (1956) Principles of Embryology. Allen & Unwind: London, UK.
Wallner K, Li GC (1986) Adriamycin resistance, heat resistance and radiation response in Chinese hamster fibroblasts. Int J Radiat Oncol Biol Phys 12: 829–833.
Xia L, Zhang D, Du R, Pan Y, Zhao L, Sun S, Hong L, Liu J, Fan D (2008) MiR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int J Cancer 123: 372–379.
Xue Z, Yan H, Li J, Liang S, Cai X, Chen X, Wu Q, Gao L, Wu K, Nie Y, Fan D (2011) Identification of cancer stem cells in vincristine preconditioned SGC7901 gastric cancer cell line. J Cell Biochem 113: 302–312.
Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, Velculescu VE, Kinzler KW, Vogelstein B, Iacobuzio-Donahue CA (2010) Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467: 1114–1117.
Yamada T, Takaoka AS, Naishiro Y, Hayashi R, Maruyama K, Maesawa C, Ochiai A, Hirohashi S (2000) Transactivation of the multidrug resistance 1 gene by T-cell factor 4/beta-catenin complex in early colorectal carcinogenesis. Cancer Res 60: 4761–4766.
Zhou JX, Aliyu MDS, Aurell E, Huang S (2012) Quasi-potential landscape in complex multi-stable systems. J R Soc Interf 9: 3539–3553.
Zhou JX, Pisco AO, Qian H, Huang S (2014) Nonequilibrium population dynamics of phenotype conversion of cancer cells. PLoS ONE 9: e110714.
Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M, Nakauchi H, Sorrentino BP (2001) The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med 7: 1028–1034.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Pisco, A., Huang, S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: ‘What does not kill me strengthens me’. Br J Cancer 112, 1725–1732 (2015). https://doi.org/10.1038/bjc.2015.146
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.146
Keywords
This article is cited by
-
Biophysical control of plasticity and patterning in regeneration and cancer
Cellular and Molecular Life Sciences (2024)
-
Dormancy, stemness, and therapy resistance: interconnected players in cancer evolution
Cancer and Metastasis Reviews (2023)
-
The role of host response to chemotherapy: resistance, metastasis and clinical implications
Clinical & Experimental Metastasis (2023)
-
Bladder cancer cells shift rapidly and spontaneously to cisplatin-resistant oxidative phosphorylation that is trackable in real time
Scientific Reports (2022)
-
Cancer: More than a geneticist’s Pandora’s box
Journal of Biosciences (2022)
