
Abstract
Background:
The phosphoinositide-3 kinase (PI3K) pathway is an attractive therapeutic target. However, difficulty in predicting therapeutic response limits the clinical implementation of PI3K inhibitors. This study evaluates the utility of clinically relevant magnetic resonance imaging (MRI) biomarkers for noninvasively assessing the in vivo response to the dual PI3K/mTOR inhibitor BEZ235 in two ovarian cancer models with differential PI3K pathway activity.
Methods:
The PI3K signalling activity of TOV-21G and TOV-112D human ovarian cancer cells was investigated in vitro. Cellular and vascular response of the xenografts to BEZ235 treatment (65 mg kg−1, 3 days) was assessed in vivo using diffusion-weighted (DW) and dynamic contrast-enhanced (DCE)-MRI. Micro-computed tomography was performed to investigate changes in vascular morphology.
Results:
The TOV-21G cells showed higher PI3K signalling activity than TOV-112D cells in vitro and in vivo. Treated TOV-21G xenografts decreased in volume and DW-MRI revealed an increased water diffusivity that was not found in TOV-112D xenografts. Treatment-induced improvement in vascular functionality was detected with DCE-MRI in both models. Changes in vascular morphology were not found.
Conclusions:
Our results suggest that DW- and DCE-MRI can detect cellular and vascular response to PI3K/mTOR inhibition in vivo. However, only DW-MRI could discriminate between a strong and weak response to BEZ235.
Similar content being viewed by others

Main
Ovarian cancer is the leading cause of death from gynaecological cancer in the western world (Siegel et al, 2012). For over 30 years, the standard treatment for ovarian cancer has been platinum-based chemotherapy in combination with, for example, taxane, or more recently liposomal doxorubicin (Vaughan et al, 2011). Although the majority of patients respond well to first-line therapy, the median progression-free survival is only 18 months (McGuire et al, 1996) and the patients often relapse with chemotherapy-resistant cancer (Vaughan et al, 2011). Hence, new therapeutic approaches are urgently needed to improve patient survival (Vaughan et al, 2011; Banerjee and Kaye, 2013).
Ovarian cancers, like other cancers, often show a deregulation of the phosphoinositide 3-kinase (PI3K) signalling pathway (Zhang et al, 2003; Brader and Eccles, 2004; Chalhoub and Baker, 2009), which plays an important role in the regulation of tumour growth, angiogenesis, metabolism and metastasis (Brader and Eccles, 2004). Hence, the PI3K pathway has been proposed as an important target for ovarian cancer treatment, especially in patients who are resistant to chemotherapy (Yap et al, 2009; Santiskulvong et al, 2011; Carden et al, 2012). Currently, the efficacy of PI3K inhibitors cannot be predicted based on gene mutation profiles (Rahman et al, 2012). It is therefore necessary to identify new biomarkers for early monitoring of response to treatment and/or development of therapeutic resistance.
At present, treatment response is assessed by measuring levels of the serum marker CA 125 (Rustin et al, 2011) and by assessing tumour burden according to the Response Evaluation Criteria in Solid Tumours (RECIST) guidelines using CT (Eisenhauer et al, 2009). However, these criteria have limited predictive value when assessed early during treatment (Ferrandina et al, 2008). In addition, measuring change in tumour volume might not always be applicable for molecularly targeted drugs, where response is often associated with functional (e.g., vascular or cytological) rather than volumetric changes (Desar et al, 2009; Serkova, 2011; Tunariu et al, 2012). This highlights a need for novel noninvasive biomarkers for assessment of early response in order to allow customisation of treatment for nonresponders.
Currently, magnetic resonance imaging (MRI) is not used for monitoring ovarian cancer treatment in the clinic, but it may be a suitable choice as it is noninvasive and provides functional imaging biomarkers (Vargas et al, 2013). Diffusion-weighted MRI (DW-MRI) can be used to measure the apparent diffusion coefficient (ADC) of water molecules, and has been proposed as a marker for tissue cellularity (Hamstra et al, 2007; Thoeny and Ross, 2010). Treatment-induced cell death can be reflected by increased ADC values even before significant tumour volume changes occur (Hamstra et al, 2007; Kyriazi et al, 2011), and DW-MRI is therefore suggested as a method for measuring early treatment response (Padhani et al, 2009). Dynamic contrast-enhanced MRI (DCE-MRI) investigates the vascularisation of tissue by measuring signal enhancement curves after intravenous administration of a contrast agent and can be used to measure changes in tumour blood flow, vascular permeability and extracellular extravascular and vascular volumes (Leach et al, 2003; O'Connor et al, 2012). The DCE-MRI has been used to assess tumour response to various drugs and has also been proposed as a tool for measuring response to PI3K pathway inhibitors (Schnell et al, 2008; Maira et al, 2009).
In this study we used two human ovarian tumour cell lines derived from different clinical phenotypes: a clear-cell carcinoma (TOV-21G) and an endometrioid carcinoma (TOV-112D) (Provencher et al, 2000). We first showed higher PI3K pathway activity in TOV-21G cells compared with TOV-112D cells. We then tested the cellular and vascular response of xenograft tumours to the dual PI3K/mTOR inhibitor BEZ235 (Maira et al, 2008) using DW- and DCE-MRI, respectively. In addition, immunohistochemistry was used to confirm cellular response, and ex vivo micro-CT (μCT) angiography was performed to investigate treatment-induced changes in vascular morphology. The main aim of this study was to compare the performance of DW-MRI and DCE-MRI for monitoring early response to PI3K inhibition.
Materials and methods
Cell lines
Human ovarian cancer cell lines TOV-21G (ATCC CRL-11730) and TOV-112D (ATCC CRL-11731) were purchased directly from ATCC (Manassas, VA, USA). The cells were grown in a 1 : 1 mixture of Medium 199 (41150; Gibco, Life Technologies, Oslo, Norway) and MCDB 105 Medium (M6395; Sigma-Aldrich, Oslo, Norway) supplemented with 15% fetal bovine serum and 0.05 mg ml−1 Gentamicin. Cells were grown under sterile conditions in 5% CO2 and 95% air at 37 °C and used before they reached 20 passages.
PI3K signalling activity in cell extracts
The PI3K signalling activity in the TOV-21G and TOV-112D was determined by immunoblotting. The cells were lysed in urea buffer (8 M urea, 0.5% Triton X, 0.1 M DTT, 1 × complete protease inhibitor (11873580001; Roche Diagnostics, Oslo, Norway), 2% (V/V) PIC 2/3). Proteins were separated by SDS–polyacrylamide electrophoresis, electrotransferred to a polyvinylidene difluoride membrane and the membranes were developed with antibodies for total Akt, phosphorylated Akt (pAkt; Ser473), phosphorylated S6 ribosomal protein (pS6; Ser235/236) and total S6 ribosomal protein (Cell Signaling Technology, Leiden, Netherlands; cat. nos. 2920s, 4060, 4858 and 2317s). The amount of β-actin (ab6276; Abcam, Cambridge, UK) in the extracts was used as control of protein loading. Binding of the primary antibodies was detected using secondary antibodies labelled with NIR fluorescent dyes. The images from the Odyssey Infrared Imaging System were processed using Image Studio 3.1 software (LI-COR Biosciences, Lincoln, NE, USA).
Tumour model and treatment
All experimental procedures involving animals were approved by The National Animal Research Authority (Application ID 4423), and carried out according to the European Convention for the Protection of Vertebrates used for Scientific Purposes.
Female athymic BalbC nu/nu mice (Taconic Tornbjerg, Ejby, Denmark) were inoculated subcutaneously on the hind limb with 5 × 106 cells of either the TOV-21G or TOV-112D cell line in PBS with 1% FBS. After an average time of 3 weeks post inoculation for the TOV-112D model and 4 weeks for the TOV-21G model, the mice were randomised into treatment and control groups. BEZ235 (LC Laboratories, Woburn, MA, USA) was dissolved in dimethyl sulphoxide (DMSO, Sigma-Aldrich D-5879) and then diluted to a final concentration of 6.5 mg ml−1 using polyethylene glycol (PEG300, 20237-1; Sigma-Aldrich). The treatment groups received BEZ235 at a dose of 65 mg kg−1 per day by oral gavage on three consecutive days. This treatment regime was expected to lead to measurable treatment response (e.g., reduced pAKT levels) in responding tumours (Maira et al, 2008; Santiskulvong et al, 2011; Moestue et al, 2013). The control groups did not receive any treatment. The study design is visualised in Figure 1.

Study design showing group sizes, treatment time points and imaging time points. (A) Magnetic resonance imaging (MRI) was performed for all mice on day 0 after which they were randomly assigned to treatment and control groups. The mice in the treatment group received 65 mg kg−1 BEZ235 on three consecutive days (‘Tx’ indicates administration of drug). After the last treatment on day 3, the mice were imaged again using MRI after which they were killed and the tumours were excised for histology. (B) For μCT the mice were randomly assigned to treatment and control groups and treated in the same way as the mice that underwent MRI. After the treatment on day 3, the mice were perfused with Microfil and the tumours were harvested for ex vivo μCT.
In vivo magnetic resonance imaging and analysis
In vivo MRI was performed on a 7T Bruker Biospec (Bruker Biospin, Ettlingen, Germany) with a 72 mm volume resonator for RF transmission and a quadrature mouse brain surface coil for reception. Both xenograft models (each n=4 control, n=8 treated) were imaged 1 day before treatment (day 0) and 2 to 6 h after the last treatment (day 3). The mice were anaesthetised with isoflurane (∼2% in 67% N2 and 33% O2) during imaging, the respiration rate was monitored and the body temperature was kept at 37 °C by circulating warm water.
Five sagittal slices with a field of view=23 × 23 mm2, slice thickness=0.692 mm and interslice distance=1 mm were acquired. On day 3, the centre slice was matched as closely as possible to the centre slice of the day 0 scans.
Anatomical MRI
High-resolution T2-weighted (T2w) anatomical images were acquired using a rapid acquisition relaxation-enhanced (RARE) spin echo (SE) sequence with echo time (TE)=12 ms, repetition time (TR)=2000 ms, RARE factor=8, number of averages (NA)=4 and matrix size=256 × 256. Tumour regions of interest (ROI) were drawn manually and downsampled to the resolution of the DW and DCE images. The tumour volume was computed according to V=1/6 π × d1 × d2 × d3, where d1, d2 and d3 are three orthogonal diameters measured from the anatomical MR images.
Diffusion-weighted MRI
Diffusion-weighted images were acquired using SE-echo planar imaging (EPI): TE=28 ms, TR=3000 ms, 4 segments, NA=4, matrix size=64 × 64, b-values=100, 300, 600 and 1000 s mm−2, diffusion gradient separation time=14 ms, diffusion gradient duration=7 ms, three orthogonal gradient orientations and fat suppression.
The ADC maps were computed using in-house software (PE Goa, NTNU) developed in MATLAB (Mathworks Inc., Natick, MA, USA). For each voxel, the logarithm of the signal intensity vs b-values was fitted linearly to obtain ADC values for each gradient direction, followed by averaging over the three gradient directions to obtain mean ADC maps. The median ADC for each tumour was calculated for the entire tumour ROI. Skewness and kurtosis of the ADC distributions for individual tumours were computed. In addition, ADC histograms of the pooled tumours for each group were created with bin size 100 × 10−6 mm2 s−1.
Dynamic contrast-enhanced MRI
Precontrast T1 values were measured voxel wise by acquiring a series of images with varying TR using a RARE SE sequence with TE=7.2 ms, TR=150, 750, 1500, 2500, 4500 and 12 000 ms, RARE factor=2, matrix size =64 × 64. This was followed by a series of 200 T1w images acquired using a RARE spin echo sequence with TE=7.2 ms, TR=300 ms, RARE factor=4 and matrix size=64 × 64. After 10 baseline scans, a bolus dose of 0.3 mmol kg−1 gadodiamide (Omniscan, GE Healthcare, Oslo, Norway) in saline was injected into the tail vein within 4 s.
For every voxel in the dynamic scans we calculated the signal intensity 1 min after contrast agent injection relative to the baseline signal intensity (RSI1 min) and the area under the signal-enhancement curve for the first minute after contrast agent injection (AUC1 min). Voxels with RSI1 min <50% and/or AUC1 min <0 were excluded from the tumour ROI for further DCE analysis as slowly or nonenhancing voxels do not fulfil the conditions for two-compartment modelling (Tofts, 1997). The precontrast T1 maps were used to convert the signal-intensity-time curves from the dynamic images to contrast-agent-time curves for each voxel as described in Jensen et al (2010). Assuming a two-compartment model (Tofts et al, 1999) and a population-based bi-exponential input function (Jensen et al, 2007), the volume transfer constant (Ktrans), the extravascular extracellular space per unit volume of tissue (ve) and the blood plasma volume fraction (vp) were estimated voxel wise by curve fitting in MATLAB. For each tumour we calculated the median Ktrans, ve, vp, RSI1 min and AUC1 min and the fraction of enhancing voxels with RSI1 min >50% (FEV1 min).
One TOV-21G tumour had unsuccessful contrast agent injection and was excluded from the DCE analysis and one TOV-112D tumour received too much contrast agent that was accounted for in the data modelling but RSI1 min and AUC1 min values were excluded.
Histology
After the MR examination on day 3, tumours were excised, fixed in 4% formaldehyde for at least 72 h, cut in half along the plane of MR imaging and embedded in paraffin. For each cell line, sections from two treated and two untreated xenografts (4 μm thick, two to three sections per tumour) were stained with haematoxylin–eosin–saffron (HES) and immunohistochemistry for pAKT or Ki-67 expression was performed. The procedures were performed for all sections at the same time and with the same protocol so that pre-analytical conditions were comparable for all samples. For pAKT labelling, the sections were pretreated with 1 × Target Retrieval Solution pH9 (S2367; Dako, Oslo, Norway) for 10 min using jet stream microwaves. Then, the sections were blocked using Odyssey blocking buffer (927-40000; Li-COR Biosciences) diluted 1 : 1 in Tris-buffered saline with 0.1% Tween (TBST) for 40 min at room temperature. The sections were incubated with rabbit anti-pAktser473 antibody (4060; Cell Signaling Technology) diluted 1 : 250 in Odyssey blocking buffer/TBST 1 : 1 overnight at 4 °C. Subsequently, the slides were incubated with donkey anti-rabbit antibody conjugated with IR-680 dye (926–68073, LI-COR Biosciences) diluted 1 : 1000 in Odyssey blocking buffer/TBST 1 : 1 for 40 min at room temperature. Negative control sections were prepared by incubating only with the secondary antibody. Near-infrared immunofluorescent imaging was performed as described previously (Moestue et al, 2013). The pAktser473 staining intensity was measured using Image Studio 3.1. The ROIs of the tumour tissue were drawn while avoiding necrotic tissue using adjacent HES-stained sections as reference. The mean signal intensity for each tumour was determined and the mean signal from adjacent negative control sections was subtracted to correct for nonspecific antibody labelling.
The Ki-67 staining was performed using an automated staining machine (Dako Autostainer). The sections were rinsed with wash buffer (S3006, Dako) and then blocked using endogenous peroxidase block (S2023, Dako) for 10 min. The sections were incubated with anti-Ki-67 antibody (SP6) (ab16667; Abcam), diluted 1 : 50 in antibody diluent (K8006, Dako) for 40 min, rinsed and then incubated with horseradish peroxidase (HRP)-labelled polymer (K4011, Dako) for 30 min at room temperature. Subsequently, the sections were incubated with DAB substrate (K4011, Dako) for two 5 min cycles. Finally, the sections were counterstained with haematoxylin. Cell morphology and Ki-67 staining of treated and control xenografts were visually compared for multiple fields of view of the tumour sections. Only nuclear staining was considered Ki-67 positive. Staining intensity was not assessed.
Ex vivo imaging of tumour vasculature and analysis
Xenografts of another cohort of mice underwent ex vivo high-resolution μCT angiography (TOV-21G: n=4 control and n=4 treated; TOV-112D: n=3 control and n=3 treated) as described previously (Kim et al, 2011). Briefly, the mice were first perfused with the contrast agent Microfil (FlowTech Inc., Carver, MA, USA), and high-resolution μCT of the xenografts was performed at 8 μm isotropic resolution. The μCT vasculature was segmented using a multi-step procedure developed by us recently (Cebulla et al, 2014) and, additionally, peritumoural tissue was masked using co-registered high-resolution MR images. The tumour fractional blood volume (FBV), mean vessel density, median vessel radii and median branch lengths were calculated. Experimental details for tumour preparation, μCT and data analysis can be found in Supplementary Methods S1.
Statistical analysis
Statistical analysis was performed using SPSS software version 19.0 (IBM Corporation, Amonk, NY, USA). An independent sample t-test was used to compare the treatment and control groups where equal variances were assumed if Levene’s test was positive for equal variances, otherwise nonequal variances were assumed. A paired sample t-test was used to assess longitudinal changes between day 0 and day 3 of the experiment within the treatment or control group. The level of significance was defined as P<0.05. Summarising statistics in the text are given as mean percent change from baseline±s.d.
Results
The PI3K pathway is constitutively activated in TOV-21G cells but not in TOV-112D cells in vitro
A clearly elevated level of the phosphorylated, active form of Akt, a key downstream effector of PI3K, was found in TOV-21G cells compared with TOV-112D cells (Figure 2A). Phosphorylation of the ribosomal protein S6 represents one of the end points of the PI3K pathway. Consistent with an elevated PI3K signalling, the level of phosphorylated S6 was elevated in the TOV-21G cells compared with the TOV-112D cells (Figure 2B).

Levels of activated Akt are elevated in TOV-21G cells and decrease in xenografts after BEZ235 treatment, whereas the levels are low in TOV-112D cells and unchanged in xenografts by treatment. (A) Immunoblots show that levels of pAkt (Ser473) are higher in cell extracts from untreated TOV-21G compared with TOV-112D. The level of β-actin served as a control for protein loading. (B) Levels of p-S6 (Ser 235/236) and total S6 in cell extracts from untreated cells are higher in TOV-21G cells compared with TOV-112D. Numbers indicate relative protein levels adjusted for loading. (C) Immunostaining of tissue sections shows that pAkt levels are lower in BEZ235-treated TOV-21G xenografts compared with controls. The TOV-112D cells have low levels in pAkt that are unchanged by treatment. Adjacent tumour sections for each tumour served as negative (neg.) controls and displayed a very low signal intensity. The scale bars represent 1 mm.
BEZ235 decreases pAktser473 levels in TOV-21G xenografts
To confirm that BEZ235 affects the PI3K pathway in vivo, we measured levels of pAkt in xenograft sections (Figure 2C). At baseline, mean pAktser473 levels were ∼4 times higher for the TOV-21G xenografts (mean±mean deviation: 3.81±0.45 a.u.) compared with TOV-112D xenografts (0.97±0.03 a.u.). There was no difference in pAktser473 levels between control (0.97±0.03 a.u.) and treated (1.01±0.19 a.u.) TOV-112D xenografts. In contrast, treated TOV-21G xenografts (2.03±0.25 a.u.) had two times lower levels of pAktser473 compared with control xenografts (3.81±0.45 a.u.).
PI3K/mTOR inhibition reduces cell proliferation and tumour growth
Examination of the Ki-67-stained sections from the TOV-21G xenografts showed a markedly reduced fraction of proliferating cells in treated TOV-21G but not TOV-112D xenografts (Figure 3A). Similarly, HES-stained tumour sections displayed a looser cellular structure and cytoplasmic vacuolisation in the treated TOV-21G xenografts, and may reflect degenerative changes in the tumour tissue caused by treatment. In both models, untreated xenografts showed an increase in tumour volume within 3 days. Treatment resulted in slightly slower tumour growth for the TOV-112D xenografts compared with the control group. In contrast, treated TOV-21G xenografts decreased in volume, leading to a significant difference in tumour growth between the treated and control groups (Figure 3B). Table 1 shows the change in tumour volume as well as MRI-derived parameters. A more detailed table including baseline and day 3 absolute values is Supplementary Table S1.

Immunohistochemistry and changes in tumour volume and ADC indicate a cellular response to treatment of the TOV-21G xenografts. (A) Haematoxylin–eosin–saffron (HES) staining of tumour sections visualises an overall lighter cytoplasmic stain for the treated TOV-21G xenograft compared with the control, whereas there is no obvious difference between treated and control TOV-112D xenografts. In the high-magnification image, the tissue of the TOV-21G xenograft shows cytoplasmic vacuolisation and a looser structure than the control tissue. The Ki-67 staining of TOV-21G xenografts indicates that both cancer and stromal cells are highly proliferating. In treated xenografts, the fraction of proliferating cells appears markedly reduced. In TOV-112D xenografts, there is no conspicuous difference in the fraction of proliferating cells between treated and control xenografts. The images were taken at × 4 and × 40 magnification and the scale bars represent 1 mm and 0.1 mm, respectively. The × 40 fields were captured at ∼0.5 mm distance from the tumour rim. (B) Change in relative tumour volume for treatment (tx) and control (ctrl) groups of both TOV-21G and TOV-112D xenografts. (C and D) Pooled histograms of ADC values in the treatment and control TOV-21G groups for day 0 and day 3. Independent sample t-test BEZ235 vs control: ###P<0.001. Paired sample t-test day 0 vs day 3: **P<0.01.
DW-MRI reveals increased ADC in TOV-21G xenografts after treatment
Treatment significantly increased (+19.3±4.8%) the median ADC for the treated TOV-21G xenografts between day 0 and day 3. All other groups did not show significant changes in median ADC (Table 1). Histogram analysis was performed to investigate changes in ADC distribution. For the treated TOV-21G xenografts, a shift of the pooled histogram from low to medium ADC values could be observed (Figure 3C), consistent with an increase in median ADC. The pooled histograms for day 0 and day 3 look similar for the untreated TOV-21G xenografts (Figure 3D) and the TOV-112D xenografts (not shown). The histogram skewness and kurtosis increased for the untreated TOV-21G xenografts, whereas both parameters decreased significantly for the treated TOV-21G xenografts (Table 1). The TOV-112D xenografts showed a large variation in skewness and kurtosis changes and no differences between control and treated groups were detected (Table 1).
Representative pre- and post-treatment ADC maps illustrate that the ADC values increased in TOV-21G tumours with some observable heterogeneity (Figure 4E and F). Central necrotic areas, which appear red in the ADC map and light pink in the HES-stained histology section (Figure 4Q), did not expand or change significantly but a general increase in ADC of the tumour section was found. For the TOV-112D tumours, there was little change in ADC over the treatment period (Figure 4G and H).

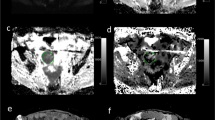
Parametric maps of a TOV-21G and TOV-112D xenograft of the same animal before and after treatment. (A–D) Anatomical images; (E–H) ADC maps; (I–L) ve maps; (M–P) Ktrans maps; (Q and R) HES-stained sections matching the post-treatment MRI parametric maps. The maps show that ADC and ve increased throughout the tumour after treatment in the TOV-21G xenografts but not in TOV-112D xenografts. Ktrans did not change in either tumour.
DCE-MRI indicates treatment-induced changes in vascular function in TOV-21G and TOV-112D xenografts
The TOV-21G xenografts exhibited a decrease (−26±10%) in ve in control tumours over 3 days and a large increase (+102±55%) in ve induced by treatment with BEZ235. For the TOV-112D xenografts the same trend was observed but the changes were smaller (−12±9% for control and +29±24% for treated tumours; Table 1). The parametric maps illustrate this differential response between the two tumour types (Figure 4I–L). Combining the ADC and ve data from the TOV-21G model, BEZ235-treated tumours could be clearly differentiated from control tumours. For the TOV-112D tumours only a slight separation was found based on differences in change in ve (Figure 5).

Treated TOV-21G xenografts can be well separated from the control group based on changes in median ADC and ve. Change in median ve vs change in median ADC for control and treatment groups of TOV-21G and TOV-112D xenografts.
There was an increase in AUC1 min, RSI1 min and FEV1 min for treated tumours and a decrease for control tumours derived from both cell lines during the 3-day treatment resulting in significant differences between treated and control groups (Table 1 and Figure 6A and B). Median vp and Ktrans did not exhibit significant differences between treated and control groups for either tumour type (Table 1). However, it should be noted that changes in Ktrans were mostly positive for the treated tumours and negative for the control tumours of both types (Figure 6C).

Treatment-induced increases in AUC1 min, FEV1 min and Ktrans indicate a change in vascular function in both TOV-21G and TOV-112D xenografts. (A) Change in AUC1 min, (B) change in FEV1 min and (C) change in Ktrans for treatment and control groups of both TOV-21G and TOV-112D xenografts. Independent sample t-test BEZ235 (Tx) vs control: #P<0.05, ##P<0.01. Paired sample t-test day 0 vs day 3: *P<0.05.
No changes in vascular morphology were detected by μCT
Tumour vascularisation was investigated in treated and untreated xenografts using μCT angiography. The parameters tumour FBV, median vessel branch length, median vessel radius and mean tumour vessel density were derived from the μCT data (Figure 7). No significant differences were detected in any of the parameters between the TOV-21G and TOV-112D xenografts or between treated and control groups. The tumour vascularisation was heterogeneous in all groups. Vessel density and FBV exhibited large variability within each group. The visual appearance of blood vessels in the tumours also varied between and within the groups.

Data on vascular morphology derived from μ CT showing no significant differences in vascular morphology between treated and control groups. (A–D) Mean values±s.d. of FBV, mean vessel density, median vessel length and median radius for treated and untreated TOV-112D (n=3 each) and TOV-21G (n=4 each) xenografts. (E) Maximum intensity projection of a 2-mm-thick tumour slice of the raw μCT data of a TOV-112D xenograft.
Discussion
In this study, the cellular and vascular response to the dual PI3K/mTOR inhibitor BEZ235 was investigated in two ovarian cancer types. Both DW-MRI and DCE-MRI were evaluated as in vivo tools for noninvasive monitoring of response to PI3K/mTOR inhibition.
The two ovarian cancer cell lines used in this study exhibit different PI3K pathway activation profiles in vitro. While the TOV-21G cells show a highly active PI3K pathway as measured by high levels of pAkt and pS6, the pathway is less active in the TOV-112D cell line. Importantly, our in vivo data showed a strong treatment response for the TOV-21G xenografts as the tumour volume decreased significantly, whereas for the TOV-112D xenografts only a slight growth suppression was detected. In addition, decreased pAkt levels in the treated TOV-21G xenografts and low pAkt levels in TOV-112D xenografts, which were unchanged by treatment, support a classification of the TOV-21G xenografts as ‘strongly responsive’ and the TOV-112D xenografts as ‘weakly responsive’ to PI3K/mTOR inhibition.
Although our data indicate a correlation between PI3K signalling activity and sensitivity for targeted therapy, other studies have failed to identify a clear correlation between drug sensitivity and PI3K mutation status in ovarian cancer (Rahman et al, 2012). In addition, determination of the mutation status of tumours is challenging. First, biopsies need to be taken, which is a highly invasive procedure. Second, tumours are spatially heterogeneous, meaning that a limited number of small tissue biopsies may not accurately represent tumour polyclonality. Third, the absence of known activating mutations does not exclude pathway hyperactivity, and vice versa – presence of activating mutations identified by DNA sequencing seems to not always cause activation of the pathway (López-Knowles et al, 2010). Therefore, the use of noninvasive, 3D imaging methods that can confirm response to therapy in patients selected for treatment with PI3K inhibitors and other drugs is highly desirable.
Several studies have previously reported that BEZ235 reduces cell proliferation and induces cell death in tumour cells in the form of apoptosis or autophagy, which in most cases led to tumour growth inhibition or shrinkage (Brachmann et al, 2009; Liu et al, 2009; Santiskulvong et al, 2011; Hong et al, 2014). Degenerative changes with cytoplasmic vacuolisation are also found in our histological data, suggesting that DW-MRI may be a suitable method to study cellular changes resulting from PI3K/mTOR inhibition. Indeed, in our study we found ADC to be a useful biomarker for response to BEZ235. In the strongly responsive TOV-21G xenografts, this parameter effectively distinguished the BEZ235-treated tumours from the untreated controls. In contrast, no significant difference between treated and control tumours was found in the weakly responsive TOV-112D xenografts. Our results indicate that median ADC values as well as ADC skewness and kurtosis may be useful in the assessment of in vivo response to PI3K/mTOR inhibition in ovarian xenografts, similar to how these parameters have been shown to detect early response to chemotherapy in ovarian cancer patients (Kyriazi et al, 2011).
Another approach for monitoring response to therapy in cancer is the use of DCE-MRI, which provides information on tumour perfusion and vascular permeability. The parameter ve is a measure of the extravascular extracellular space and, similar to ADC, may be related to the cellular density of the tumour tissue. In a recent study, Sampath et al (2013) showed that response to the PI3K/mTOR inhibitor GDC-0980 was associated with increased ve. In our study, ve was a good marker for discriminating between treated and control xenografts derived from both cancer cell lines. This is in contrast to tumour volume and ADC, which only showed significant differences between treated and control groups of the strongly responsive TOV-21G tumours.
Besides its importance for cellular functions, the PI3K pathway has been shown to influence angiogenesis via several mechanisms, for example, via hypoxia-inducible factor 1, nitric oxide or angiopoietins as summarised by Brader and Eccles (2004) and Karar and Maity (2011). Several groups have extensively studied the effects of PI3K/mTOR inhibitors on vascular morphology and function in preclinical tumour models using multiple imaging techniques, but report varying results. For example, Qayum et al (2009) have investigated the vascular effects of the PI3K/mTOR inhibitor PI-103 and found increased blood flow, vascular density, branch length and reduced tortuosity. Similar results were found with BEZ235 in a second study by this group (Fokas et al, 2012). The same group then investigated the delivery of doxorubicin into xenografts after treatment with the specific PI3Kα/δ inhibitor GDC-0941 and found increased doxorubicin delivery and, as in the previous studies, increased blood flow (Qayum et al, 2012). Collectively, they interpreted these vascular changes as ‘vascular normalisation’. In contrast, Sampath et al (2013) reported ‘antivascular effects’ such as increased vessel size and decreased FBV, vessel density, blood flow and Ktrans after treatment with a PI3K inhibitor (GNE-490) or a PI3K/mTOR inhibitor (GDC-0980). In yet another study by Schnell et al (2008), response to BEZ235 was associated with reduced Ktrans, and also decreased capillary permeability.
In contrast to previous studies (Qayum et al, 2009; Fokas et al, 2012; Sampath et al, 2013), our μCT data did not show treatment-induced changes in vascular morphology at the time points we investigated. The xenografts exhibited a heterogeneous vascularisation, with some tumours exhibiting large areas filled with contrast agent, which could be a result of contrast agent leaking out of brittle vessels. This may have caused the large spread in FBV. However, the mean vessel density, median branch length and median diameter were not affected by this and still did not show significant vascular differences between the groups.
Despite no change in vascular morphology as measured by μCT, we did find changes in vascular function using DCE-MRI. All parameters describing early tumour enhancement (i.e., AUC1 min, RSI1 min and FEV1 min) increased, which is associated with increased delivery of contrast agent, and may be a sign for improved perfusion. This compares favourably to the studies by Qayum et al (2009, 2012) and Fokas et al (2012) in which increased tumour perfusion and increased delivery of doxorubicin into the tumour was observed. This enhanced perfusion and contrast agent extravasation may partly be attributable to the decreased interstitial fluid pressure commonly found in solid tumours (Hompland et al, 2012). In addition, the increase we found in Ktrans may be a sign for improved vascular function. In contrast, Schnell et al (2008) and Sampath et al (2013) reported decreasing Ktrans. This discrepancy may be caused by the varying effect that PI3K inhibitors can have on the vasculature ranging from antivascular to vascular normalising, which can influence changes in Ktrans. In addition, Ktrans is dependent on both vascular perfusion and permeability, making this parameter difficult to interpret. Importantly, the treatment-induced vascular changes detected by non-model-based DCE parameters were more significant compared with those detected by the model-based parameter Ktrans.
In our study we used xenografts with very different PI3K pathway activities and an interesting finding was that the changes in vascular function did not seem to depend on the activity of the PI3K pathway.
In light of the above-mentioned studies, including ours, we postulate that changes in vascular morphology and function induced by PI3K/mTOR inhibition can take various forms and are likely dependent on the dosage and mechanism of action of the drug, as well as the characteristics of the tumour model employed. Vascular changes following PI3K/mTOR inhibition are therefore not only difficult to predict but also difficult to compare across studies when different imaging methods are used. In contrast, DW-MRI is a relatively well-established method for measuring cellular density. Its use may be especially encouraged because it a fast imaging modality that does not require contrast agent administration or complex post-processing such as advanced pharmacokinetic modelling to provide quantitative data. In addition, compared with DCE-MRI, DW-MRI is probably less dependent on image acquisition and quantification methods, making it attractive for clinical applications. However, a standardisation of imaging protocols and data analysis is still needed (Padhani et al, 2009). Recently, other studies have reported that DW-MRI is useful for the assessment of treatment response to other targeted drugs (Beloueche-Babari et al, 2013; Williams et al, 2013).
Currently, several PI3K inhibitors are evaluated in ovarian cancer, partly because of a connection between chemoresistance and the activity of the PI3K pathway (Carden et al, 2012). Concomitantly, therapy monitoring will be crucial for determining drug dosages and therapeutic efficacy to avoid unnecessary side effects in nonresponsive patients. The DW-MRI is not commonly employed for assessing response to targeted therapies, and to our knowledge no other studies investigating the response to PI3K inhibition using DW-MRI have been published. Our study suggests that DW-MRI may be a highly relevant method for noninvasive monitoring of response to PI3K inhibitors in ovarian cancer, as we demonstrated that increased ADC and a change in histogram-based parameters are associated with inhibition of tumour growth. Incorporation of DW-MRI into clinical trials could be done easily and would be necessary in order to define clinically relevant time points at which a potential change in ADC parameters can be seen (Padhani et al, 2009). DCE-MRI, which provides complementary information on vascular changes, may also be a suitable technique for evaluating the efficacy of PI3K inhibitors. However, in this study, DCE-MRI could not discriminate between a strong and weak response in the xenografts. On the other hand, this discrimination could be performed with DW-MRI, indicating that DW-MRI holds a strong potential in the assessment of treatment response to PI3K inhibitors. As this technique is less dependent on fitting mathematical models to the imaging data and does not require use of contrast agents, it may be the preferred method for clinical therapy monitoring.
Change history
03 February 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Banerjee S, Kaye SB (2013) New strategies in the treatment of ovarian cancer: current clinical perspectives and future potential. Clin Cancer Res 19: 961–968.
Beloueche-Babari M, Jamin Y, Arunan V, Walker-Samuel S, Revill M, Smith PD, Halliday J, Waterton JC, Barjat H, Workman P, Leach MO, Robinson SP (2013) Acute tumour response to the MEK1/2 inhibitor selumetinib (AZD6244, ARRY-142886) evaluated by non-invasive diffusion-weighted MRI. Br J Cancer 109: 1562–1569.
Brachmann SM, Hofmann I, Schnell C, Fritsch C, Wee S, Lane H, Wang S, Garcia-Echeverria C, Maira SM (2009) Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proc Natl Acad Sci USA 106: 22299–22304.
Brader S, Eccles SA (2004) Phosphoinositide 3-kinase signalling pathways in tumor progression, invasion and angiogenesis. Tumori 90: 2–8.
Carden CP, Stewart A, Thavasu P, Kipps E, Pope L, Crespo M, Miranda S, Attard G, Garrett MD, Clarke PA, Workman P, de Bono JS, Gore M, Kaye SB, Banerji U (2012) The association of PI3 kinase signaling and chemoresistance in advanced ovarian cancer. Mol Cancer Ther 11: 1609–1617.
Cebulla J, Kim E, Rhie K, Zhang J, Pathak AP (2014) Multiscale and multi-modality visualization of angiogenesis in a human breast cancer model. Angiogenesis 17: 695–709.
Chalhoub N, Baker SJ (2009) PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol 4: 127–150.
Desar IM, van Herpen CM, van Laarhoven HW, Barentsz JO, Oyen WJ, van der Graaf WT (2009) Beyond RECIST: molecular and functional imaging techniques for evaluation of response to targeted therapy. Cancer Treat Rev 35: 309–321.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45: 228–247.
Ferrandina G, Ludovisi M, Corrado G, Carone V, Petrillo M, Scambia G (2008) Prognostic role of Ca125 response criteria and RECIST criteria: analysis of results from the MITO-3 phase III trial of gemcitabine versus pegylated liposomal doxorubicin in recurrent ovarian cancer. Gynecol Oncol 109: 187–193.
Fokas E, Im JH, Hill S, Yameen S, Stratford M, Beech J, Hackl W, Maira SM, Bernhard EJ, McKenna WG, Muschel RJ (2012) Dual inhibition of the PI3K/mTOR pathway increases tumor radiosensitivity by normalizing tumor vasculature. Cancer Res 72: 239–248.
Hamstra DA, Rehemtulla A, Ross BD (2007) Diffusion magnetic resonance imaging: a biomarker for treatment response in oncology. J Clin Oncol 25: 4104–4109.
Hompland T, Ellingsen C, Ovrebo KM, Rofstad EK (2012) Interstitial fluid pressure and associated lymph node metastasis revealed in tumors by dynamic contrast-enhanced MRI. Cancer Res 72: 4899–4908.
Hong SW, Shin JS, Moon JH, Kim YS, Lee J, Choi EK, Ha SH, Lee DH, Chung HN, Kim JE, Kim KP, Hong YS, Lee JL, Lee WJ, Choi EK, Lee JS, Jin DH, Kim TW (2014) NVP-BEZ235, a dual PI3K/mTOR inhibitor, induces cell death through alternate routes in prostate cancer cells depending on the PTEN genotype. Apoptosis 19: 895–904.
Jensen LR, Berge K, Bathen TF, Wergedahl H, Schonberg SA, Bofin A, Berge RK, Gribbestad IS (2007) Effect of dietary tetradecylthioacetic acid on colon cancer growth studied by dynamic contrast enhanced MRI. Cancer Biol Ther 6: 1810–1816.
Jensen LR, Huuse EM, Bathen TF, Goa PE, Bofin AM, Pedersen TB, Lundgren S, Gribbestad IS (2010) Assessment of early docetaxel response in an experimental model of human breast cancer using DCE-MRI, ex vivo HR MAS, and in vivo 1H MRS. NMR Biomed 23: 56–65.
Karar J, Maity A (2011) PI3K/AKT/mTOR pathway in angiogenesis. Front Mol Neurosci 4: 51.
Kim E, Zhang J, Hong K, Benoit NE, Pathak AP (2011) Vascular phenotyping of brain tumors using magnetic resonance microscopy (muMRI). J Cereb Blood Flow Metab 31: 1623–1636.
Kyriazi S, Collins DJ, Messiou C, Pennert K, Davidson RL, Giles SL, Kaye SB, Desouza NM (2011) Metastatic ovarian and primary peritoneal cancer: assessing chemotherapy response with diffusion-weighted MR imaging—value of histogram analysis of apparent diffusion coefficients. Radiology 261: 182–192.
Leach MO, Brindle KM, Evelhoch JL, Griffiths JR, Horsman MR, Jackson A, Jayson G, Judson IR, Knopp MV, Maxwell RJ, McIntyre D, Padhani AR, Price P, Rathbone R, Rustin G, Tofts PS, Tozer GM, Vennart W, Waterton JC, Williams SR, Workman P (2003) Assessment of antiangiogenic and antivascular therapeutics using MRI: recommendations for appropriate methodology for clinical trials. Br J Radiol 76 Spec No 1: S87–S91.
Liu TJ, Koul D, LaFortune T, Tiao N, Shen RJ, Maira SM, Garcia-Echevrria C, Yung WK (2009) NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol Cancer Ther 8: 2204–2210.
López-Knowles E, O'Toole SA, McNeil CM, Millar EKA, Qiu MR, Crea P, Daly RJ, Musgrove EA, Sutherland RL (2010) PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int J Cancer 126: 1121–1131.
Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, Garcia-Echeverria C (2008) Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther 7: 1851–1863.
Maira SM, Stauffer F, Schnell C, Garcia-Echeverria C (2009) PI3K inhibitors for cancer treatment: where do we stand? Biochem Soc Trans 37: 265–272.
McGuire WP, Hoskins WJ, Brady MF, Kucera PR, Partridge EE, Look KY, Clarke-Pearson DL, Davidson M (1996) Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med 334: 1–6.
Moestue SA, Dam CG, Gorad SS, Kristian A, Bofin A, Maelandsmo GM, Engebraten O, Gribbestad IS, Bjorkoy G (2013) Metabolic biomarkers for response to PI3K inhibition in basal-like breast cancer. Breast Cancer Res 15: R16.
O'Connor JP, Jackson A, Parker GJ, Roberts C, Jayson GC (2012) Dynamic contrast-enhanced MRI in clinical trials of antivascular therapies. Nat Rev Clin Oncol 9: 167–177.
Padhani AR, Liu G, Koh DM, Chenevert TL, Thoeny HC, Takahara T, Dzik-Jurasz A, Ross BD, Van Cauteren M, Collins D, Hammoud DA, Rustin GJ, Taouli B, Choyke PL (2009) Diffusion-weighted magnetic resonance imaging as a cancer biomarker: consensus and recommendations. Neoplasia 11: 102–125.
Provencher DM, Lounis H, Champoux L, Tetrault M, Manderson EN, Wang JC, Eydoux P, Savoie R, Tonin PN, Mes-Masson AM (2000) Characterization of four novel epithelial ovarian cancer cell lines. In Vitro Cell Dev Biol Anim 36: 357–361.
Qayum N, Im J, Stratford MR, Bernhard EJ, McKenna WG, Muschel RJ (2012) Modulation of the tumor microvasculature by phosphoinositide-3 kinase inhibition increases doxorubicin delivery in vivo. Clin Cancer Res 18: 161–169.
Qayum N, Muschel RJ, Im JH, Balathasan L, Koch CJ, Patel S, McKenna WG, Bernhard EJ (2009) Tumor vascular changes mediated by inhibition of oncogenic signaling. Cancer Res 69: 6347–6354.
Rahman M, Nakayama K, Rahman MT, Nakayama N, Ishikawa M, Katagiri A, Iida K, Nakayama S, Otsuki Y, Shih IeM, Miyazaki K (2012) Clinicopathologic and biological analysis of PIK3CA mutation in ovarian clear cell carcinoma. Hum Pathol 43: 2197–2206.
Rustin GJ, Vergote I, Eisenhauer E, Pujade-Lauraine E, Quinn M, Thigpen T, du Bois A, Kristensen G, Jakobsen A, Sagae S, Greven K, Parmar M, Friedlander M, Cervantes A, Vermorken J Gynecological Cancer I (2011) Definitions for response and progression in ovarian cancer clinical trials incorporating RECIST 1.1 and CA 125 agreed by the Gynecological Cancer Intergroup (GCIG). Int J Gynecol Cancer 21: 419–423.
Sampath D, Oeh J, Wyatt SK, Cao TC, Koeppen H, Eastham-Anderson J, Robillard L, Ho CC, Ross J, Zhuang G, Reslan HB, Vitorino P, Barck KH, Ungersma SE, Vernes JM, Caunt M, Van Bruggen N, Ye W, Vijapurkar U, Meng YJ, Ferrara N, Friedman LS, Carano RA (2013) Multimodal microvascular imaging reveals that selective inhibition of class I PI3K is sufficient to induce an antivascular response. Neoplasia 15: 694–711.
Santiskulvong C, Konecny GE, Fekete M, Chen KY, Karam A, Mulholland D, Eng C, Wu H, Song M, Dorigo O (2011) Dual targeting of phosphoinositide 3-kinase and mammalian target of rapamycin using NVP-BEZ235 as a novel therapeutic approach in human ovarian carcinoma. Clin Cancer Res 17: 2373–2384.
Schnell CR, Stauffer F, Allegrini PR, O'Reilly T, McSheehy PM, Dartois C, Stumm M, Cozens R, Littlewood-Evans A, Garcia-Echeverria C, Maira SM (2008) Effects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 on the tumor vasculature: implications for clinical imaging. Cancer Res 68: 6598–6607.
Serkova NJ (2011) Translational imaging endpoints to predict treatment response to novel targeted anticancer agents. Drug Resist Updat 14: 224–235.
Siegel R, Naishadham D, Jemal A (2012) Cancer statistics, 2012. CA Cancer J Clin 62: 10–29.
Thoeny HC, Ross BD (2010) Predicting and monitoring cancer treatment response with diffusion-weighted MRI. J Magn Reson Imaging 32: 2–16.
Tofts PS (1997) Modeling tracer kinetics in dynamic Gd-DTPA MR imaging. J Magn Reson Imaging 7: 91–101.
Tofts PS, Brix G, Buckley DL, Evelhoch JL, Henderson E, Knopp MV, Larsson HB, Lee TY, Mayr NA, Parker GJ, Port RE, Taylor J, Weisskoff RM (1999) Estimating kinetic parameters from dynamic contrast-enhanced T(1)-weighted MRI of a diffusable tracer: standardized quantities and symbols. J Magn Reson Imaging 10: 223–232.
Tunariu N, Kaye SB, Desouza NM (2012) Functional imaging: what evidence is there for its utility in clinical trials of targeted therapies? Br J Cancer 106: 619–628.
Vargas HA, Barrett T, Sala E (2013) MRI of ovarian masses. J Magn Reson Imaging 37: 265–281.
Vaughan S, Coward JI, Bast RC Jr., Berchuck A, Berek JS, Brenton JD, Coukos G, Crum CC, Drapkin R, Etemadmoghadam D, Friedlander M, Gabra H, Kaye SB, Lord CJ, Lengyel E, Levine DA, McNeish IA, Menon U, Mills GB, Nephew KP, Oza AM, Sood AK, Stronach EA, Walczak H, Bowtell DD, Balkwill FR (2011) Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer 11: 719–725.
Williams TM, Galban S, Li F, Heist KA, Galban CJ, Lawrence TS, Holland EC, Thomae TL, Chenevert TL, Rehemtulla A, Ross BD (2013) DW-MRI as a predictive biomarker of radiosensitization of GBM through targeted inhibition of checkpoint kinases. Transl Oncol 6: 133–142.
Yap TA, Carden CP, Kaye SB (2009) Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer 9: 167–181.
Zhang L, Yang N, Katsaros D, Huang W, Park JW, Fracchioli S, Vezzani C, Rigault de la Longrais IA, Yao W, Rubin SC, Coukos G (2003) The oncogene phosphatidylinositol 3'-kinase catalytic subunit alpha promotes angiogenesis via vascular endothelial growth factor in ovarian carcinoma. Cancer Res 63: 4225–4231.
Acknowledgements
Animals were housed in the Comparative Medicine Core Facility, Norwegian University of Science and Technology (NTNU). In vivo MRI was performed at the MR Core Facility, NTNU. Paraffin embedding, sample sectioning and HES staining were performed at the CMIC Core Facility, NTNU. Ex vivo MRI was performed at the BRB Molecular Imaging Service Center, The Johns Hopkins University. Micro-CT imaging was performed by Numira Biosciences (Salt Lake City, UT, USA). We thank Elise Sandsmark for help with the acquisition of images of HES-stained tumour sections. The research was supported by a Leiv Eiriksson mobility stipend (224788/F11) from the Norwegian Research Council and a Travel and Research Grant from the Norwegian Research School in Medical Imaging (MedIm) to JC; student grants to AvdV from the Dutch VSB fonds and Prins Bernhard cultuurfonds (Klok Korsmit Tacke fonds); a Susan G. Komen for the Cure Career Catalyst Grant (KG090640) to APP; a grant from the Norwegian Cancer Society and the Norwegian Breast Cancer Society (2209215-2011) to SAM; grants from the Norwegian Cancer Society (171656–PR-2009-0270) and The Cancer Research Foundation at St Olav’s University Hospital to GB.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Cebulla, J., Huuse, E., Pettersen, K. et al. MRI reveals the in vivo cellular and vascular response to BEZ235 in ovarian cancer xenografts with different PI3-kinase pathway activity. Br J Cancer 112, 504–513 (2015). https://doi.org/10.1038/bjc.2014.628
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.628
Keywords
This article is cited by
-
Mechanism of anticancer effect of ETP-45658, a PI3K/AKT/mTOR pathway inhibitor on HT-29 Cells
Medical Oncology (2023)
-
Diffusion-weighted imaging and diffusion kurtosis imaging for early evaluation of the response to docetaxel in rat epithelial ovarian cancer
Journal of Translational Medicine (2018)
-
Use of non-invasive imaging to monitor response to aflibercept treatment in murine models of colorectal cancer liver metastases
Clinical & Experimental Metastasis (2017)
-
Dactolisib (NVP-BEZ235) toxicity in murine brain tumour models
BMC Cancer (2016)
