
Abstract
Background:
p53 is a transcription factor with tumour suppressor properties, which is able to induce mitochondrial apoptosis independently of its transcriptional activity. We recently synthesised two new compounds (ISA27 and SM13), which block p53-MDM2 interaction and induce apoptosis in p53 wild-type (WT) tumour cells. The aim of this study was to verify the effectiveness of these compounds in tumours carrying a mutated form of p53 gene with no transcriptional activity.
Methods:
In vitro we evaluated the effectiveness of our compounds in cancer cell lines carrying WT, mutated and null p53 gene. In vivo study was performed in Balb/c nude mice and the mitochondrial-dependent apoptotic signalling was evaluated by western blot.
Results:
Both ISA27 and SM13 reduced cell proliferation and induced apoptosis in vitro in cells carrying either p53 WT or mutated gene, suggesting that its effect is independent from p53 transcriptional activity. On the contrary, SM13 had no effect in a p53 null cell line. In vivo, ISA27 and SM13 induced cancer cell death in a dose-dependent manner through the activation of the mitochondrial-dependent death signalling in p53-mutated cells. In vivo, SM13 reduced tumour growth.
Conclusions:
Our study proposes SM13 as anticancer compound to use for the treatment of p53-dependent tumours, even in the absence of p53 transcriptional activity.
Similar content being viewed by others

Main
p53 is best known as a transcription factor that binds to specific DNA sequences and transactivates a number of genes involved in the regulation of cell cycle arrest and apoptosis (Riley et al, 2008). In addition to this nuclear activity, p53 also possesses transcription-independent biological activities that take place far from the nucleus. Indeed, the overexpression of a mutant p53, lacking most of the DNA-binding domain and completely deficient in transactivation function, can efficiently induce apoptosis in human cells (Haupt et al, 1997; Kakudo et al, 2005). Under a variety of cell-death-inducing conditions, p53 rapidly moves to mitochondria where it induces mitochondrial outer membrane permeabilisation, thereby triggering the release of pro-apoptotic factors from the mitochondrial intermembrane space (Kroemer et al, 2007).
p53 family protein activity is regulated in a complex way, including posttranslational modifications, protein stabilisation, protein–protein interaction and modulation of subcellular localisation (Kim et al, 2002; Harms et al, 2004; Bourdon et al, 2005; Harms and Chen, 2005).
In particular, p53 binds MDM2, an ubiquitin ligase that ubiquitinates p53 protein and causes inactivation, nuclear export and degradation of p53 (Momand et al, 1992; Haupt et al, 1997; Geyer et al, 2000). In turn, p53 enhances the expression of MDM2, although MDM2 levels can be also regulated via p53-independent pathways (Phelps et al, 2003). Therefore, MDM2 induction in response to p53 is the major negative feedback loop aimed at blocking p53 proapoptotic function and thus allowing cell survival (Malaguarnera et al, 2007). As most of human cancers are p53-dependent owing to inactivating mutations of p53 gene, overexpression of its inhibitors, reduction of its activators or inactivation of its downstream targets (Green and Kroemer, 2009), the possibility to disrupt the MDM2-p53 interaction to enhance p53-dependent inhibition of cancer cell proliferation and survival, has become an attractive goal for cancer therapy (Brown et al, 2009). In the last years, several potent small molecules, which disrupt MDM2-p53 interaction, have been developed and one of them, Nutlin-3, has entered early phase clinical trials (Shangary and Wang, 2009). In the clinical scenario, different cancers express different levels of p53 and also p53 mutant variants, leading to resistance to p53-targeting compounds. On the basis of these findings, we recently synthesised two new compounds:(3R,7aR)-6-(4-chlorobenzyl)-1H-spiro[imidazo[1,5-c]thiazole-3,3′-indoline]-2′,5,7(6H,7aH)-trione (9c) (Gomez-Monterrey et al, 2010), named ISA27, and 5-bromo-3’-(cyclohexane carbonyl)-1-methyl-2-oxospiro[indoline-3,2’-thiazolidine] (4n) (Bertamino et al, 2013), named SM13. These compounds are able to induce apoptosis of human tumour cell lines both in vitro and in vivo (Gomez-Monterrey et al, 2010; Bertamino et al, 2013; Costa et al, 2013). SM13, in particular, is a promising anticancer compound, which has proven to be more effective than Nutlin at lower doses in different tumour cell lines (Bertamino et al, 2013). We have demonstrated that SM13 inhibits MDM2-induced p53 degradation and activates apoptotic events in vitro in human tumour cells carrying a wild-type (WT) p53 gene (Bertamino et al, 2013). So far, its efficacy has never been tested in vivo. Furthermore, the efficacy of these compounds has never been tested in tumours bearing p53 variants-lacking transcriptional activity. It is possible to speculate that in these cancers, p53 accumulation can still be effective to inhibit cancer growth by enhancing mitochondrial apoptosis. The present study was therefore undertaken to verify whether SM13 and ISA27 retain effectiveness in cancer cells with mutated p53. Also, as SM13 was never tested in vivo, we sought to verify whether the compound is as efficient in vivo as in vitro.
Materials and Methods
Cell culture
MCF7, which express a WT variant of p53, KAT-4, which bears a p53 variant mutated at codon 273 (CGT→CAT; Arg→His), BHT101, which expresses a p53 mutant at codon 251 (ATC→ACC; Ile→Thr) and FRO cells, which do not express p53 at all, were cultured in Dulbecco’s minimal essential medium (DMEM) supplemented with 10% foetal bovine serum (FBS) at 37 °C in 95% air – 5%CO2.
Compounds
Lyophilised ISA27 and SM13 were dissolved in absolute DMSO. Intra-peritoneal and intra-tumour injections of absolute DMSO were used for the treatment of control mice.
Immunoprecipitation and western blot
Immunoprecipitation and western blot analysis were performed as described previously (Iaccarino et al, 2006; Sorriento et al, 2008, 2009). Anti-p53, MDM2, Bax, Citochrome c, Caspase 9 and Actin antibodies were from Santa Cruz Biotechnology, Inc. (Heidelberg, Germany); anti-cleaved caspase 3 antibody was from Cell Signalling (Danvers, MA, USA).
Cell proliferation and DNA synthesis assay
Cell proliferation and DNA synthesis assays were performed as described previously (Santulli et al, 2011).
Tunel assay
Apoptosis was evaluated in KAT-4 cells after treatment with SM13, using the DeadEnd Colorimetric Tunel System from Promega (Madison, WI, USA), following the manufacturer’s instructions. Results are expressed as mean±s.d. of apoptotic nuclei.
In vivo study design
Experiments were carried out, in accordance to NIH guidelines for Animal Investigation, in 6-week-old BALB/c nude mice (Charles River Italia, Calco, Italy), which had access to food and water ad libitum. For tumour formation, a suspension containing 2 × 106 KAT-4 cells in 200 μl of DMEM were injected subcutaneously in the dorsal side of nude mice, as previously described (Sorriento et al, 2009). Animals were anesthetised using isofluorane 2%. We used mice that developed tumours of ∼6 mm in diameter by 2 weeks. Mice were divided into four groups (5 mice per group) and administered twice a week for 2 weeks with intra-tumour or intra-peritoneal injections (IP) of the specific treatment (ISA27 and SM13).
In particular, two groups received intra-tumour injection of ISA27 or SM13 either at low (low: 1 mg kg–1) or high dosages (high: 3 mg kg–1); another group received IP of 5 mg kg–1 of the compounds and; the control group received intra-tumour or IP of DMSO. The IP of DMSO did not modify tumour growth with respect to intra-tumour injection, thus in figures we include intra-tumour injection of DMSO as control for treated tumours. Tumour growth was measured by caliper twice a week and expressed as tumour volumes in mm3 according to the formula ‘Volume=(width)2 × length/2’. At the end of the treatment, mice were killed by cervical dislocation and tumours were processed for biochemical or histological analysis. The Federico II University Ethical Committed for Animal Studies approved all in vivo experimental protocols.
Real-time PCR
Total RNA from tumours was isolated using Trizol reagent (Invitrogen, Life Technologies, Grand Island, NY, USA) and cDNA was synthetised by means of Thermo-Script real-time polymerase chain reaction (RT-PCR) System (Invitrogen, Life Technologies), following the manufacturer’s instruction. After reverse transcription reaction, RT quantitative PCR was performed with the SYBR Green RT PCR master mix kit (Applied Biosystems, Life Technologies), as described previously (Ciccarelli et al, 2011). Primers for cytokines gene analysis were as follows:
TNFα: forward, 5′-CCAGGAGAAAGTCAGCCTCCT-3′ and reverse 5′-CGATAAAGGGGTCAGAGTAAT-3′;
VEGF: forward, 5′-CAGGCTGTCGTAACGATGAA-3′ and reverse 5′-TTTCTTGCGCTTTCGTTTTT-3′;
MMP-9: forward, 5′-CGTCGTGATCCCCACTTACT-3′and reverse 5′-AACACACAGGGTTTGCCTTC-3′;
IL-1β: forward, 5′-GCCTTGGGCCTCAAAGGAAAGAAT-3′ and reverse 5′-GGAAGACACAGATTCCATGGTGAAG-3′;
p53: forward, 5′-TGAACCGGAGGCCCATCCTC-3′ and reverse, 5′-GGCACAAACACGCACCTCAAA-3′;
p21: forward, 5′-CCTGGGACCTCACCTGCTCTGCTG-3′ and reverse, 5′-GCAGAAGATGTAGAGCGGGCCTTT-3′;
GADD45: forward, 5′-TGCTCAGCAAAGCCCTGAGT-3′ and reverse, 5′-GCAGGCACAACACCACGTTA-3′.
All values obtained were normalised to the values obtained with the 18S primers. The results are expressed as the relative integrated intensity.
Histology and Immunocytochemistry
Paraffin-embedded sections were processed for the triple-layered immunocytochemical peroxidase antiperoxidase method (Sorriento et al, 2012). PCNA (Sigma Aldrich, Milano, Italy) and Cleaved caspase 3 (Abcam, Cambridge, UK) antiserum were used to analyze cell proliferation and death, respectively. The peroxidase was revealed in presence of 0.03% hydrogen peroxide and of an electron donor, 2.5% diaminobenzidine, which becomes visible as a brown precipitate. For negative controls, the primary antiserum was omitted. Sections were then viewed with an Eclipse E1000 Fluorescence Microscope (Nikon, Milan, Italy) and acquired using Sigma Scan Pro software (Jandel), as described previously (Cittadini et al, 2009). For toxicity assay, Masson trichrome staining was performed in paraffined sections from the liver, kidney and lung of mice treated with IP of ISA27 or SM13 and control mice, as previously described (Santulli et al, 2012).
Statistical analysis
All values are presented as mean±s.e.m. Two-way ANOVA was performed to compare the different parameters among the different groups. A significance level of P<0.05 was assumed for all statistical evaluations. Statistics were computed with GraphPad Prism Software (San Diego, CA, USA).
Results
Effects of ISA27 in different tumour cell lines
We evaluated the effect of ISA27 on p53 levels and activation of caspase 3 in MCF-7 (carrying p53 WT gene), BHT-101 and KAT-4 cells (carrying p53 mutated gene). In all these cell lines, ISA27 increases both p53 and cleaved caspase 3 levels (Figure 1A), suggesting that it inhibits MDM2-dependent p53 degradation and increases apoptotic events in all cell lines. As ISA27 exerts the same effect in p53 WT (MCF7) and p53-mutated cells (KAT-4 and BHT-101) and given the hotspot mutation of p53 within the DNA-binding domain in KAT-4 and BHT-101 cells, which inhibits its transcriptional activity (Blagosklonny et al, 1998), the effect of ISA27 in the regulation of apoptosis should be independent from p53 transcriptional activity. We therefore performed the study in KAT-4 cells, which represent a well-established model for cancer studies in our laboratory (Sorriento et al, 2009). We first confirmed the effectiveness of ISA27 to disrupt MDM2/p53 binding in KAT-4 cells by co-immunoprecipitation assay. Supplementary Figure S1A shows that MDM2-precipitated p53 and ISA27 reduced this phenomenon. ISA27-dependent increase of p53 levels is due to the inhibition of protein degradation rather than the induction of p53 gene expression. Indeed, p53 gene expression levels, evaluated by RT PCR, were unchanged in treated cells compared with controls (Supplementary Figure S1B). To evaluate the effect of ISA27 on tumour cell growth, we studied in vitro cell proliferation indicators such as cell number and DNA synthesis. ISA27 inhibits both cell proliferation (Figure 1B) and DNA synthesis (Figure 1C) in a time-dependent manner. This finding suggests that ISA27 is able to induce p53-dependent apoptosis also in KAT-4 cells harboring the p53 variant.

Effects of ISA27 on tumour cell growth in vitro. (A) The effect of ISA 27 on p53 and cleaved caspase 3 levels were evaluated in whole lysates from different tumour cell lines by western blot. In all MCF-7, BHT-101 and KAT-4 cells, the treatment with ISA 27 increases p53 levels and consequently leads to an increase of apoptotic signalling. Actin was used as loading control. Images are representative of three independent experiments. (B, C) KAT-4 cells were treated with ISA27 for 24 and 48 h and cell proliferation was analyzed (B). ISA27 reduces cell proliferation in a time-dependent manner. Results are the mean of five independent experiments and are presented as mean±s.e.m. This effect was confirmed by analysis of DNA synthesis by means of tritiated thymidine incorporation (C). DNA synthesis was reduced by ISA27 treatment (*P<0.05 vs control 24H; **P<0.05 vs control 48H). Results are the mean of five independent experiments and are presented as mean±s.e.m.
Effects of ISA27 on KAT-4 cell growth in vivo
To confirm in vitro data, we tested the effect of ISA27 in vivo. In BALB/c nude mice, the injection of 2 × 106 KAT-4 cells in the dorsal lateral region results in the development of a ∼6-mm diameter tumour in 2 weeks, in about 70% of mice. Tumours were treated with different doses of ISA27 by means of IP or intra-tumour injection, as described in Materials and Methods. The effect of ISA27 on tumour growth appears to be dose-dependent. Indeed, intra-tumour injection of high doses (3 mg kg–1) leads to a progressive regression of tumour, whereas low doses (1 mg kg–1) or intra-peritoneal treatment (5 mg kg–1) can only delay tumour growth (Figure 2A). At the end of the treatment, mice were killed and tumours were taken for biochemical and histological analysis. We first evaluated p53 and MDM2 levels by western blot to confirm the effectiveness of the treatment in vivo. The p53 levels were increased in all treated tumours (Figure 2B). Also, MDM2 levels were increased in treated tumours with respect to control (Figure 2B), probably as a compensative response to the increased levels of p53. We then evaluated apoptosis and proliferation in tumours by western blot. Figure 2B shows that ISA27 increased cleaved caspase 3 levels and reduced the phosphorylation of RB in a dose-dependent manner. These results were confirmed by immunohistochemical analysis in paraffin-embedded sections (Figure 3A and B). ISA27-treated tumours show dose-dependent increase of cleaved caspase 3 levels (Figure 3A) and a reduction of PCNA expression, marker of cell proliferation (Figure 3B). These data confirm that ISA27 induces apoptosis also in vivo in a dose-dependent manner.

Effects of ISA27 on tumour growth in vivo. (A) To validate our in vitro results, we studied the effects of ISA27 in a cancer model in vivo. Tumour growth was measured twice a week by a caliper during all the treatment long (14 days of treatment). Intra-peritoneal treatment (IP: 5 mg Kg–1) or low doses (LOW: 1 mg Kg–1) of ISA27 retard tumour growth compared with controls. High doses (HIGH: 3 mg Kg–1) are more effective, significantly reducing tumour size. Results are the mean of measurements from five mice per group (*P<0.05 vs control). The upper panel shows a representative image of tumours at the end of the treatment. (B) Tumours were homogenized and MDM2, p53, active caspase 3, and RB levels were analyzed. Actin was used as control. ISA27 induces an increase of p53 and cleaved caspase 3 levels and a reduction of p-RB expression in treated tumours compared with controls. MDM2 level were unchanged in treated tumours with respect to controls. Images are representative of three independent experiments.

Histological analysis of ISA27-treated tumours. (A, B) 14 days from starting treatment, mice were killed and tumours were taken for histological analysis. Cell death and proliferation were evaluated by analysis of cleaved caspase 3 and PCNA levels by immunohistochemistry in paraffin-embedded sections of tumours. ISA27-treated tumours show a dose-dependent increased cleaved caspase 3 levels (B) and reduced cell proliferation (A). Images are representative of three independent experiments.
Effects of SM13 on KAT-4 cell proliferation in vitro
The effectiveness of the nutlin-derivate compound, SM13, in the regulation of apoptotic signalling in a p53 WT tumour cell line (MCF-7) has already been demonstrated. Here we evaluated its effects also in a p53 mutant cell line, KAT-4. We first confirmed the effectiveness of SM13 to disrupt MDM2/p53 binding by co-immunoprecipitation assay. Supplementary Figure S2A shows that MDM2 precipitated p53 and SM13 reduced this phenomenon. We then tested the effect of SM13 on tumour cell growth in vitro. SM13 reduced KAT-4 cell proliferation both at 24 and 48 h from starting treatment (Figure 4A). Giving the mutation of p53 gene in KAT-4 cells, which inhibits its transcriptional activity, we evaluated the effect of SM13 on mitochondrial-dependent apoptotic signalling. Indeed, recent studies have shown that interactions of p53 with various members of the Bcl-2 family cause mitochondrial-mediated apoptosis in a transcription-independent manner (Speidel, 2010; Ha et al, 2013). Accordingly, our data showed that SM13 induced p53 levels, increased the expression of the proapoptotic protein Bax, leading to the release of cytochrome c (Figure 4B). This event leads to the activation of both Caspase 9 and Caspase 3, thus, inducing cell death (Figure 4B). Accordingly, also the levels of Cyt-c in cytosolic extracts were increased (Supplementary Figure S2B). We have showed above that ISA27 induced an increase of p53 levels by inhibiting protein degradation without modifying its gene expression levels. We demonstrated that also SM13 did not modify gene expression of p53 (Supplementary Figure S2C), thus confirming that SM13 rather inhibits p53 degradation. To confirm the apoptotic effect of SM13, we evaluated DNA fragmentation through a Tunel assay. Images show that treatment with SM13 induced apoptotic events in tumour cells (Figure 4C). All together, these data suggest that SM13 is able to induce apoptosis in tumour cells by activating mitochondrial apoptotic signalling. To assess the specificity of SM13 effectiveness in the regulation of p53-dependent apoptosis, we evaluated its effects in FRO cells, a tumour cell line which does not express p53 (Wolf and Rotter, 1985; Namba et al, 1995). Figure 4D shows that SM13 increased cleaved caspase 3 levels in KAT-4, but not in FRO cells; thus, suggesting that the ability of SM13 to induce apoptosis strictly depends on p53. Moreover, to confirm the p53 transcription independent effect of SM13, we evaluated gene expression of p53 target genes, p21 and Gadd45. In a p53 WT tumour cell line, MCF7, the treatment with SM13 induced p21 and Gadd45 gene expression, whereas in p53-mutant cell type, KAT-4, such phenomenon was reduced (Figure 4E). These data suggest that the effects of SM13 are strictly dependent on p53, and given the lack of transcriptional activity of p53 (Figure 4E), they are due to the activation of p53-dependent mitochondrial apoptotic pathway.

Effects of SM13 on tumour cell growth in vitro. (A) KAT-4 cells were treated with SM13 for 24 and 48 h, and cell proliferation was analyzed. SM13 reduced cell proliferation in a time-dependent manner (*P<0.05 vs control 24H; **P<0.05 vs control 48H). Results are representative of five independent experiments and are presented as mean±s.e.m. (B) To evaluate the mechanism of action of SM13, we analyzed its effect on mitochondrial-dependent apoptotic signalling by western blot. SM13-dependent increase of p53 induces activation of Bax, release of citochrome c from mitochondria and activation of caspase 9 and caspase 3. Images are the mean of three independent experiments. (C) To confirm the effect of SM13 on apoptosis, we evaluated DNA fragmentation through a TUNEL assay. Positive nuclei were counted and results were expressed in graph as mean±s.d. SM13 is able to induce apoptotic events in tumour cells. Images are representative of three independent experiments. (D) The ability of SM13 to induce apoptosis was evaluated in FRO cells, a tumour cell line, which do not express p53. p53 and cleaved caspase 3 levels were evaluated by western blot. SM13 was not able to regulate cleaved caspase 3 levels in FRO cells with respect to KAT-4 cells. Results are representative of three independent experiments. (E) To confirm the p53 transcription-independent effect of SM13, we evaluated gene expression of p53 target genes, p21 and Gadd45. In a p53 WT tumour cell line, MCF7, the treatment with SM13 induced p21 and Gadd45 gene expression, whereas in p53-mutant cell type, KAT-4, such phenomenon was reduced (*P<0.05 vs control). Results are the mean of five independent experiments and are presented as mean±s.e.m.
Effects of SM13 on KAT-4 cell growth in vivo
To confirm in vitro data, we evaluated the effect of SM13 on tumour growth in vivo in nude mice. Tumours were treated with IP and intra-tumour injection of SM13. Data show that SM13 is a potent inhibitor of tumour growth in a dose-dependent manner (Figure 5A). Indeed, the intra-tumour injection of SM13 inhibited tumour growth at low dosages and was more efficient at high dosages. Also, the intra-peritoneal treatment strongly delayed tumour growth (Figure 5A). We compared ISA27 and SM13 effects on tumour growth and we found that at equal doses, SM13 is more effective than ISA27 to inhibit the progression of tumour (Figure 5B). Indeed, when comparing the effect on tumour growth, the three different treatment regimens consistently showed the advantage of SM13 over ISA27. Indeed, the intra-peritoneal treatment with SM13 (5 mg kg–1) more efficiently delayed tumour growth compared with ISA27 (−77±2.6% vs −56±2% at 14 days, P<0.05). Similarly, the intra tumour injection at low dosages of SM13 (1 mg kg–1) is more effective than the treatment with low dosages of ISA27 (SM13: −85±2%; ISA27: −68±3.4% at 14 days, P<0.05). Finally, the intra tumour injection of high dosages of SM13 (3 mg kg–1) induces tumour regression more efficiently than ISA27 (−98±1.6% vs −80±1.6% at 14 days, P<0.05). It was already evidenced that ISA27 and SM13 showed different minimal inhibitory concentration 50%, IC50, (SM13: 0.03 μ M±0.01; ISA27: 0.40 μ M±0.02) (Gomez-Monterrey et al, 2010; Bertamino et al, 2013) demonstrating that SM13 is 10-fold more potent than ISA27, and this is in agreement with their different effect on tumour growth at the same doses. Thus, besides the good anticancer properties of ISA27, our data show that SM13 is more efficient compared with ISA27 to inhibit tumour growth in vivo.

Effects of SM13 on tumour growth in vivo. (A) To validate in vitro results, we studied the effects of SM13 in vivo in Balb/c nude mice. Tumour growth was measured twice a week by a caliper during all the treatment long (14 days of treatment). Intra-peritoneal treatment (IP: 5 mg Kg–1) or low doses (low: 1 mg Kg–1) of SM13 retard tumour growth compared with controls and are more effective of ISA27 at the same dosages. High doses (high: 3 mg Kg–1) reduce tumour volumes back to the starting size. Results are the mean of measurements from five mice per group (*P<0.05 vs control). Figure also shows a representative image of tumours at the end of the treatment. (B) Tumours size was taken at the end of the treatment and the percent of reduction of tumour growth was calculated in tumours treated with ISA27 and SM13 with respect to controls. SM13 is able to inhibit tumour growth more efficiently than SM13 (*P<0.05 vs ISA27). (C) Tumours were homogenized to confirm by western blot analysis the effect of SM13 on mitochondrial-dependent apoptotic signalling. In treated tumours, SM13 increases protein levels of Bax and citochrome c, and activates both caspase 9 and caspase 3. Images are the mean of five independent experiments.
To confirm the mechanism by which SM13 induced apoptosis, we evaluated its effect on mitochondrial signalling in tumours from treated and control mice. According to in vitro data, intra-tumour injection of low and high doses of SM13 significantly increased p53, Bax, Cit-C, active caspase 9 and active caspase 3 levels in a dose-dependent manner (Figure 5B). Cell death and proliferation were also evaluated by immunohistochemistry analysis of cleaved caspase 3 and PCNA levels in tumours collected from control and treated mice. SM13-treated tumours show a dose-dependent reduction of cell proliferation (PCNA: Figure 6A) and an increase of apoptosis (Cleaved caspase 3: Figure 6B). To further confirm the effect of SM13 on tumour growth, we analyzed by RT PCR the expression of genes key to tumour progression and metastatisation, which are usually overexpressed in tumour cells. Figure 7 shows that VEGF (Figure 7A), MMP-9 (Figure 7B), TNFα (Figure 7C) and IL-1β (Figure 7D) expression significantly decreased in treated tumours compared with controls in a dose-dependent manner, confirming the effectiveness of the treatment to reduce tumour growth.

Histological analysis of SM13-treated tumours. (A, B) 14 days from starting treatment, mice were killed and tumours were taken for histological analysis. Cell death and proliferation were evaluated by immunohistochemical analysis of cleaved caspase 3 and PCNA levels. SM13-treated tumours show a dose-dependent increase of cleaved caspase 3 levels, marker of apoptosis (B) and reduction of PCNA, marker of cell proliferation (A). Images are representatives of three independent experiments.

Real-time PCR analysis of inflammation and angiogenesis in treated tumours. (A, D) VEGF, MMP9, TNFα and IL-1β expression was evaluated by real-time PCR to assess the ability of ISA27 to regulate pro-apoptotic factors in cancer cells. At 2 weeks from starting treatment, the expression of VEGF (A), MMP9 (B), TNFα (C) and IL-1β (D) is strongly inhibited in a dose-dependent manner (*P<0.05 vs control). Results are the mean of five independent experiments and are presented as mean±s.e.m.
Evaluation of side effects after treatment with ISA27 and SM13
Finally, we tested the safety of in vivo administration of our compounds in healthy nude mice. There were no significant changes in body weight among treated and control groups of mice (data not shown). Internal organs (liver and kidneys) were collected from controls and treated mice and morphological analysis was performed by Masson trichrome staining in paraffin-embedded sections. No morphological differences were found in tissues from treated mice compared with controls (Supplementary Figure S3). These data indicate that the treatments with ISA27 and SM13 had no effects on mice health at the doses and time of treatment that were used in this study.
Discussion
Our results show for the first time that ISA27 and SM13 are two potent inhibitors of cancer cell proliferation even in cancer cells bearing a p53 mutated-variant. p53 is a known tumour suppressor, which regulates several events, like growth arrest, senescence and apoptosis, in response to cellular damage (Levine, 1997; Wu and Levine, 1997). In basal conditions, p53 is expressed at low levels in cells owing to its continuous MDM2-dependent degradation (Levine, 1997; Wu and Levine, 1997). Rapid induction of p53 protein levels by various stress types prevents inappropriate propagation of cells carrying damaged DNA (Moll and Petrenko, 2003). p53 exerts its pro-apoptotic effect in a transcription-dependent manner in the nucleus and in a transcription independent manner in mitochondria (Chen et al, 1996; Vousden and Lu, 2002; Mihara et al, 2003).
Recently, MDM2 has emerged as the main regulator of p53 by limiting the p53 tumour suppressor function (Momand et al, 1992; Finlay, 1993; Chen et al, 1996). Indeed, small molecules have been developed recently to disrupt MDM2-p53 binding, which have been proposed as anticancer therapeutic drugs (Shangary and Wang, 2009). In this context, we have designed and synthesised a series of small molecules, which are analogues of spirooxoindolepyrrolidine nucleus and inhibit MDM2-dependent degradation of p53 by preventing p53/MDM2 binding (Gomez-Monterrey et al, 2010). Among them, ISA27 resulted the most potent compound to regulate p53 activity. We have recently demonstrated that ISA27 induces p53-dependent apoptosis in a human tumour cell line carrying a WT p53 gene (Costa et al, 2013). Here we show that such effect is reproducible also in tumour cells carrying a mutated p53 gene, which prevents p53 ability to activate transcription of target genes. On the basis of ISA27 structure, we designed and synthesised a series of new modified compounds, which should be more selective and effective than ISA27. These analogues of ISA27 were prepared to explore new structural requirements at the thiazolidine domain for the antiproliferative activity and p53 modulation. Among them, the 5-bromo-3′-(cyclohexanecarbonyl)-1-methyl-2-oxospiro[indoline-3,2′-thiazolidine]-4′-carboxylate (SM13) emerged as the most potent compound, as it inhibits cell growth in different human tumour cell lines at submicromolar and nanomolar concentrations (Bertamino et al, 2013). Docking studies confirmed the interactions of SM13 with the well-known Trp23 and Phe19 clefts, explaining the reasons for its binding affinity for MDM2. SM13 at low doses is capable of inducing the accumulation of p53 protein, inducing significant apoptotic cell death (Costa et al, 2013). Here we demonstrated that SM13, better than its precursor ISA27, is a potent inhibitor of tumour growth in cells carrying a mutated p53 gene and is already effective at low dosages. It is known that p53 induces apoptotic events through the regulation of gene transcription in the nucleus. Recently, it has been shown that this protein is also able to induce apoptosis in a transcription independent manner (Figure 8A), which is based on the activation of the mitochondrial apoptotic signalling (Arima et al, 2005). Indeed, it directly induces permeabilisation of the outer mitochondrial membrane by forming complexes with antiapoptotic proteins Bcl-xL and Bcl-2, resulting in the release of cytochrome c in the cytosol. Moreover, it has been shown that p53 translocation to mitochondria occurs earlier after TPA stimulation, leading to mitochondrial dysfunction (Zhao et al, 2005). The specific signalling involved in this transcriptional-independent effect of p53 on apoptosis is still unclear. What is known is that the transcriptional blockade of p53 by α-amanitin induces p53 mitochondrial localisation and Bax accumulation, and activation in mitochondria. Accordingly, here we show that SM13 induces apoptosis of KAT-4 cells, which express a mutated p53 protein lacking the transcriptional activity, by targeting p53-dependent mitochondrial signalling (Figure 8B). Indeed, the SM13-dependent increase of p53 levels, in KAT-4 cells is associated with the increase of BAX, release of cytochrome c, increase of cleaved caspase 3 levels and, finally, cell death. The confirmation that such mechanism strictly depends on p53, either WT or transcription activity deficient, derives from the finding that SM13 has no effect on FRO cells, a cell line that does not express p53 (Wolf and Rotter, 1985). It could be a compensatory response in those conditions, like cancer, in which p53 cannot activate gene transcription, but must induce cell death to block a pathological increase of cell growth. In conclusion, here we demonstrate that both ISA27 and SM13 are effective regulators of p53 in KAT-4 cells. Among them, SM13 seems to be the most effective as it leads to a progressive tumour regression at lower doses. In treating human tumours, a combination of selected drugs with different mechanisms of action and a low toxicity grade is preferred over monotherapy. In this context, SM13 could be a good anticancer drug to be used in combination chemotherapy, as it is selective and does not generate side effects. In this study, we demonstrated that SM13 has strong pro-apoptotic effects by regulating p53 turnover within the cell and it could be used as a prototype small molecule for cancer therapy. In conclusion, we propose SM13 as a feasible anticancer drug, which could be used alone or in combination with common anticancer agents to ameliorate the response to chemotherapy in a wide set of tumour types characterised by elevated levels of MDM2 and low levels of p53.

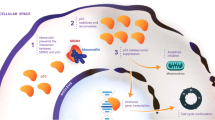
p53 transcription-dependent and -independent activation of apoptotic pathway. (A) p53 is activated in response to DNA damage and induces the apoptotic pathway through two alternative mechanisms, one dependent on transcriptional activity of p53 and one independent from transcriptional activity of p53. Indeed, activated p53 can move towards nucleus to activate the transcription of pro-apoptotic genes or move towards mitochondria to induce the release of cytochrome c. (B) Under basal conditions, MDM2 binds p53, thus leading to the degradation of the protein. SM13 inhibits MDM2/p53 binding by competing with p53 for binding to MDM2. SM13 therefore leads to the inhibition of MDM2-dependent degradation of p53 and to the increase of p53 levels. p53 binds and inhibits antiapoptotic proteins (BclXL); thus, inducing Bax levels on mitochondrial membrane, which leads to the release of citochrome c from mitochondria that on turn activates apoptotic events.
Change history
06 January 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Arima Y, Nitta M, Kuninaka S, Zhang D, Fujiwara T, Taya Y, Nakao M, Saya H (2005) Transcriptional blockade induces p53-dependent apoptosis associated with translocation of p53 to mitochondria. J Biol Chem 280 (19): 19166–19176.
Bertamino A, Soprano M, Musella S, Rusciano MR, Sala M, Vernieri E, Di Sarno V, Limatola A, Carotenuto A, Cosconati S, Grieco P, Novellino E, Illario M, Campiglia P, Gomez-Monterrey I (2013) Synthesis, in vitro, and in cell studies of a new series of [Indoline-3,2'-thiazolidine]-based p53 modulators. J Med Chem 56: 5407–5421.
Blagosklonny MV, Giannakakou P, Wojtowicz M, Romanova LY, Ain KB, Bates SE, Fojo T (1998) Effects of p53-expressing adenovirus on the chemosensitivity and differentiation of anaplastic thyroid cancer cells. J Clin Endocrinol Metab 83 (7): 2516–2522.
Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, Saville MK, Lane DP (2005) p53 isoforms can regulate p53 transcriptional activity. Genes Dev 19 (18): 2122–2137.
Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP (2009) Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer 9 (12): 862–873.
Chen J, Wu X, Lin J, Levine AJ (1996) mdm-2 inhibits the G1 arrest and apoptosis functions of the p53 tumor suppressor protein. Mol Cell Biol 16 (5): 2445–2452.
Ciccarelli M, Sorriento D, Cipolletta E, Santulli G, Fusco A, Zhou RH, Eckhart AD, Peppel K, Koch WJ, Trimarco B, Iaccarino G (2011) Impaired neoangiogenesis in beta(2)-adrenoceptor gene-deficient mice: restoration by intravascular human beta(2)-adrenoceptor gene transfer and role of NFkappaB and CREB transcription factors. Br J Pharmacol 162 (3): 712–721.
Cittadini A, Monti MG, Castiello MC, D'Arco E, Galasso G, Sorriento D, Saldamarco L, De Paulis A, Napoli R, Iaccarino G, Sacca L (2009) Insulin-like growth factor-1 protects from vascular stenosis and accelerates re-endothelialization in a rat model of carotid artery injury. J Thromb Haemost 7 (11): 1920–1928.
Costa B, Bendinelli S, Gabelloni P, Da Pozzo E, Daniele S, Scatena F, Vanacore R, Campiglia P, Bertamino A, Gomez-Monterrey I, Sorriento D, Del Giudice C, Iaccarino G, Novellino E, Martini C (2013) Human glioblastoma multiforme: p53 reactivation by a novel MDM2 inhibitor. PloS One 8 (8): e72281.
Finlay CA (1993) The mdm-2 oncogene can overcome wild-type p53 suppression of transformed cell growth. Mol Cell Biol 13 (1): 301–306.
Geyer RK, Yu ZK, Maki CG (2000) The MDM2 RING-finger domain is required to promote p53 nuclear export. Nat Cell Biol 2 (9): 569–573.
Gomez-Monterrey I, Bertamino A, Porta A, Carotenuto A, Musella S, Aquino C, Granata I, Sala M, Brancaccio D, Picone D, Ercole C, Stiuso P, Campiglia P, Grieco P, Ianelli P, Maresca B, Novellino E (2010) Identification of the Spiro(oxindole-3,3'-thiazolidine)-Based Derivatives as Potential p53 Activity Modulators. J Med Chem 53: 8319–8329.
Green DR, Kroemer G (2009) Cytoplasmic functions of the tumour suppressor p53. Nature 458 (7242): 1127–1130.
Ha JH, Shin JS, Yoon MK, Lee MS, He F, Bae KH, Yoon HS, Lee CK, Park SG, Muto Y, Chi SW (2013) Dual-site interactions of p53 protein transactivation domain with anti-apoptotic Bcl-2 family proteins reveal a highly convergent mechanism of divergent p53 pathways. J Biol Chem 288 (10): 7387–7398.
Harms K, Nozell S, Chen X (2004) The common and distinct target genes of the p53 family transcription factors. Cell Mol Life Sci 61 (7-8): 822–842.
Harms KL, Chen X (2005) The C terminus of p53 family proteins is a cell fate determinant. Mol Cell Biol 25 (5): 2014–2030.
Haupt Y, Maya R, Kazaz A, Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387 (6630): 296–299.
Iaccarino G, Izzo R, Trimarco V, Cipolletta E, Lanni F, Sorriento D, Iovino GL, Rozza F, De Luca N, Priante O, Di Renzo G, Trimarco B (2006) Beta2-adrenergic receptor polymorphisms and treatment-induced regression of left ventricular hypertrophy in hypertension. Clin Pharmacol Ther 80 (6): 633–645.
Kakudo Y, Shibata H, Otsuka K, Kato S, Ishioka C (2005) Lack of correlation between p53-dependent transcriptional activity and the ability to induce apoptosis among 179 mutant p53s. Cancer Res 65 (6): 2108–2114.
Kim SS, Chae HS, Bach JH, Lee MW, Kim KY, Lee WB, Jung YM, Bonventre JV, Suh YH (2002) P53 mediates ceramide-induced apoptosis in SKN-SH cells. Oncogene 21 (13): 2020–2028.
Kroemer G, Galluzzi L, Brenner C (2007) Mitochondrial membrane permeabilization in cell death. Physiol Rev 87 (1): 99–163.
Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88 (3): 323–331.
Malaguarnera R, Vella V, Vigneri R, Frasca F (2007) p53 family proteins in thyroid cancer. Endocr Relat Cancer 14 (1): 43–60.
Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11 (3): 577–590.
Moll UM, Petrenko O (2003) The MDM2-p53 interaction. Mol Cancer Res 1 (14): 1001–1008.
Momand J, Zambetti GP, Olson DC, George D, Levine AJ (1992) The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69 (7): 1237–1245.
Namba H, Hara T, Tukazaki T, Migita K, Ishikawa N, Ito K, Nagataki S, Yamashita S (1995) Radiation-induced G1 arrest is selectively mediated by the p53-WAF1/Cip1 pathway in human thyroid cells. Cancer Res 55 (10): 2075–2080.
Phelps M, Darley M, Primrose JN, Blaydes JP (2003) p53-independent activation of the hdm2-P2 promoter through multiple transcription factor response elements results in elevated hdm2 expression in estrogen receptor alpha-positive breast cancer cells. Cancer Res 63 (10): 2616–2623.
Riley T, Sontag E, Chen P, Levine A (2008) Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 9 (5): 402–412.
Santulli G, Basilicata MF, De Simone M, Del Giudice C, Anastasio A, Sorriento D, Saviano M, Del Gatto A, Trimarco B, Pedone C, Zaccaro L, Iaccarino G (2011) Evaluation of the anti-angiogenic properties of the new selective alphaVbeta3 integrin antagonist RGDechiHCit. J Transl Med 9: 7.
Santulli G, Cipolletta E, Sorriento D, Del Giudice C, Anastasio A, Monaco S, Maione AS, Condorelli G, Puca A, Trimarco B, Illario M, Iaccarino G (2012) CaMK4 Gene Deletion Induces Hypertension. J Am Heart Assoc 1 (4): e001081.
Shangary S, Wang S (2009) Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol 49: 223–241.
Sorriento D, Campanile A, Santulli G, Leggiero E, Pastore L, Trimarco B, Iaccarino G (2009) A new synthetic protein, TAT-RH, inhibits tumor growth through the regulation of NFkappaB activity. Mol Cancer 8: 97.
Sorriento D, Ciccarelli M, Santulli G, Campanile A, Altobelli GG, Cimini V, Galasso G, Astone D, Piscione F, Pastore L, Trimarco B, Iaccarino G (2008) The G-protein-coupled receptor kinase 5 inhibits NFkappaB transcriptional activity by inducing nuclear accumulation of IkappaB alpha. Proc Natl Acad Sci USA 105 (46): 17818–17823.
Sorriento D, Santulli G, Del Giudice C, Anastasio A, Trimarco B, Iaccarino G (2012) Endothelial cells are able to synthesize and release catecholamines both in vitro and in vivo. Hypertension 60 (1): 129–136.
Speidel D (2010) Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol 20 (1): 14–24.
Vousden KH, Lu X (2002) Live or let die: the cell's response to p53. Nat Rev Cancer 2 (8): 594–604.
Wolf D, Rotter V (1985) Major deletions in the gene encoding the p53 tumor antigen cause lack of p53 expression in HL-60 cells. Proc Natl Acad Sci USA 82 (3): 790–794.
Wu L, Levine AJ (1997) Differential regulation of the p21/WAF-1 and mdm2 genes after high-dose UV irradiation: p53-dependent and p53-independent regulation of the mdm2 gene. Mol Med 3 (7): 441–451.
Zhao Y, Chaiswing L, Velez JM, Batinic-Haberle I, Colburn NH, Oberley TD St, Clair DK (2005) p53 translocation to mitochondria precedes its nuclear translocation and targets mitochondrial oxidative defense protein-manganese superoxide dismutase. Cancer Res 65 (9): 3745–3750.
Acknowledgements
This work was supported by MIUR Grant (GI: PRIN 2009EL5WBP and PC: PRIN 2012 n° 20122ATMNJ_002) and Grant to GI from Società Italiana di Ipertensione Arteriosa.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The Authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Sorriento, D., Del Giudice, C., Bertamino, A. et al. New small molecules, ISA27 and SM13, inhibit tumour growth inducing mitochondrial effects of p53. Br J Cancer 112, 77–85 (2015). https://doi.org/10.1038/bjc.2014.577
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.577

{kind=link}
{kind=link}
{kind=link}