
Abstract
Introduction:
Previous small-molecule antiangiogenics have compromised chemotherapy dose intensity in breast cancer. We present a phase I trial of a novel selective agent, nintedanib, plus standard chemotherapy in early breast cancer.
Methods:
Her-2-negative breast cancer patients with tumours larger than 2 cm were eligible for dose-escalation trial (classic 3+3 method).
Results:
The recommended phase II dose (RP2D) was 150 mg BID of nintedanib combined with standard dose of weekly paclitaxel followed by adriamycin plus cyclophosphamide. The dose-limiting toxicity was transaminase elevation. At the RP2D, the dose intensity was ∼100%. The pathologic complete response was 50%.
Conclusions:
The combination allows the delivery of full-dose intensity, while efficacy seems promising.
Similar content being viewed by others

Main
Despite the early diagnosis screening campaigns, long-term relapse rates of HER-2-negative breast cancer range from 25% (hormone-receptor positive; Davies et al, 2013) to >40% (triple-negative breast cancer; Foulkes et al, 2010). Combinations of different cytotoxic agents or introduction of novel therapeutic schedules seem to have reached a plateau in long-term disease control (Swain et al, 2013; Earl et al, 2014). Clearly, novel therapeutic alternatives are needed.
Antiangiogenic agents are an attractive therapeutic strategy as their therapeutic effect is more dependent on the effects on the microenvironment than on the underlying oncogenic mutation (Weis and Cheresh, 2011). Breast cancer seems to be clustered into many different subtypes characterised by many non-overlapping patterns of genetic aberrations (Curtis et al, 2012; Shah et al, 2012); thus, antiangiogenic agents may be useful across several of those subtypes.
Recent randomised trials have proven increased efficacy of adding bevacizumab, a monoclonal antibody against VEGF, to standard neoadjuvant chemotherapy in HER-2-negative early breast cancer (Bear et al, 2012; von Minckwitz et al, 2012). A second class of antiangiogenic agents, small molecules with multi-tyrosine kinase inhibitor activity that target several pro-angiogenic axes (TKIs), has been studied in breast cancer as well. First-generation agents of this class, like sunitinib or sorafenib, increased the progression-free survival (Baselga et al, 2012; Bergh et al, 2012). However, in the long term, this was not translated into higher survival rates (Baselga et al, 2012; Bergh et al, 2012). One possibility accounting for these disappointing results is that these regimens were highly toxic. Second, the chemotherapy total dose intensity was significantly reduced, probably explaining the detrimental effects.
Nintedanib is a novel agent of this therapeutic class. It has activity against VEGFR1-3, PDGFRA/B, FGFR1-3 and other pro-oncogenic kinases such as RET or FTL3 in the low nanomolar range. Preclinical studies suggest a lower Km than their predecessors against pro-angiogenic kinases (Boehringer Ingelheim, GmbH, 2009). Clinical studies in combination with chemotherapy in other malignancies where other antiangiogenic drugs did not achieve improvement in overall survival suggest improved toxicity profile and long-term efficacy (Ledermann et al, 2011; Reck et al, 2014).
Thus, we aimed to determine the recommended phase II dose (RP2D) of nintedanib in combination with standard chemotherapy for neoadjuvant breast cancer and to study its tolerability during a full course of neoadjuvant chemotherapy. As a secondary objective, we describe the anti-tumour efficacy, reported as pathologic complete response. This trial was conducted with the aim of studying the efficacy of nintedanib in breast cancer in a randomised trial. The combination showed an excellent tolerance, allowing delivery of full-dose intensity, and showed an impressive preliminary activity.
Materials and Methods
Patients
Women aged >18 years with histologically confirmed HER-2-negative resectable breast cancer were eligible. Key inclusion criteria included: (1) signed informed consent form; (2) primary tumour over 2 cm on its longer diameter (measured by MRI; any nodal status and multicentricity were allowed, as long as the disease was deemed resectable by a multidisciplinary committee); (3) HER-2-negative disease; (4) ECOG 0–1; (5) adequate liver, haematologic and renal function defined by usual phase I criteria. Patients with previous treatment of any kind for breast cancer (excluding patients adequately treated >5 years ago from a previous breast cancer), undergoing hormonal replacement or contraceptives, concurrent or previous malignancy of any kind up to 5 years before diagnosis (except from non-melanoma skin cancer or in situ carcinoma of the breast or cervix), concurrent serious medical conditions, or patients undergoing anticoagulation/antiplatelet therapy (except low-dose heparin or <325 mg per day aspirin) or with a history of haemorrhagic/thromboembolic event clinically significant in the last 6 months were excluded. Patients with recent major surgery (4 weeks) were not candidates for this trial.
The study was conducted in accordance with the International Conference on Harmonisation Good Clinical Practice Guidelines, the Declaration of Helsinki and applicable local regulatory requirements and laws. The protocol was approved by the Institutional Review Board.
Study design, treatment and procedures
This was an open-label, prospective, multicentric dose-finding study. The trial was an investigator-sponsored study.
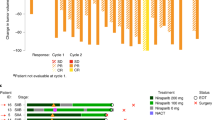
The primary objectives were to determine the RP2D and study the tolerability and safety of the combination of nintedanib with weekly paclitaxel, followed by adriamycin plus cyclophosphamide (4 × ). The secondary objective was to study the activity of the combination. The escalation followed a classic 3+3 scheme. The treatment schedule and dosages are depicted and described in Figure 1A. Two rules were considered: (1) paclitaxel, adriamycin or cyclophosphamide doses were fixed at standard doses and not to be escalated, despite of the dose level achieved for nintedanib; and (2) the maximum-tolerated dose of nintedanib in combination with paclitaxel should be below 150 mg BID, the planned randomised phase II trial would not be conducted, as according to preclinical and single-agent phase I studies, the plasma levels would be insufficient to achieve pharmacodynamic activity (Boehringer Ingelheim, GmbH, 2009; Mross et al, 2010). Neoadjuvant adriamycin plus cyclophosphamide were administered without nintedanib, due to the potential additive cardiotoxic properties.

Trial schedule. Patients received oral nintedanib twice per day during the 12 courses of weekly paclitaxel. The morning dose of nintedanib of the paclitaxel days was omitted due to previous preclinical pharmacokinetic interaction observations. One week after the last nintedanib dose (2 weeks after the last paclitaxel infusion), adriamycin plus cyclophosphamide was started, every 21 days. Surgery was programmed a minimum of 4 weeks after the last chemotherapy dose, and in any case, 5 weeks since the last nintedanib dose. Radiation and/or hormonal therapy were administered after surgery according to physician’s choice.
The primary end point was to determine the incidence and nature of dose-limiting toxicities (DLTs) of nintedanib in combination with neoadjuvant paclitaxel followed by adriamycin/cyclophosphamide graded according to NCI CTCAE v4.0 (NCI). The secondary end point was pathologic complete response measured by the Miller and Payne criteria (Ogston et al, 2003).
Patients were staged with conventional procedures; a LVEF was determined with ultrasound before registration. The patients were visited weekly during the first 3-week cycle and on days 1 and 15 subsequently, for physical and haematologic toxicity assessment. Detailed schedules for dose reductions or drug hold are provided in Supplementary Materials and Methods. The DLT evaluation period was from day 1 to 21; however, no escalation was performed until all patients in one dose level completed two cycles.
Statistical studies
Safety was evaluated in all patients who received at least one dose of study medication. Efficacy was evaluated in patients who received at least one cycle of treatment (21 days). Nonparametric test was used due to the sample size. Dose intensity was calculated as the total delivered dose divided by the planned dose, for each agent.
Results
Dose escalation, toxicity and dose delivery
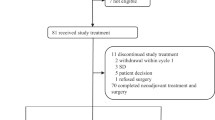
Basic patient demography is depicted in Table 1. Three patients were enroled in level 1 (150 mg BID of nintedanib); no DLTs were observed. We then escalated to level 2 (200 mg BID). One of the two patients experienced a DLT, consisting on grade 3 ALT elevation on day +8. The patient withdrew consent and came off trial. We recruited a third patient who on day +8 presented with a DLT, consisting on G4 ALT elevation plus G3 AST and GGT elevation. Nintedanib was held until recovery to <G2 (14 days later); paclitaxel was resumed 7 days earlier. The dose was reduced to level 1 and the patient continued without significant side effects. We finally recruited three more patients in dose level 1. No further DLTs were registered. Thus, 80 mg m2 of weekly paclitaxel plus 150 mg BID of nintedanib was established as the RP2D.
Besides one G3 neutropenia and two grade 3 lymphopenia, no G3 or non-tolerable G2 toxicity was reported in the 24 cycles administered in level 1. The main toxicity events are detailed in Table 2. Of note, hand–foot syndrome or blood pressure elevation, classic toxicities of this drug class, were absent or very mild, respectively, in level 1; in level 2, only one grade 3 hypertension was registered (Table 2).
The dose delivery in level one was almost complete: 99.9 and 97.4% of the planned nintedanib and paclitaxel doses were administered. No dose reductions were needed. There were two 1-week paclitaxel dose delays related to toxicity (G2 neutropenia and G1 fever, the latter deemed unrelated to study medication by the investigators).
Efficacy
Eight patients were evaluable. The pathologic complete response (breast plus axilla) was 50%. Two out of six hormone-receptor-positive patients (33%), and two out of two triple-negative patients, achieved pathologic complete response. The remainder four patients achieved G2 (three patients, 37.5%) and G3 (one patient, 12.5%) Miller and Payne response. Six out of eight patients (75%) underwent conservative surgery.
Discussion
TKIs such as sorafenib or sunitinib have been developed in breast cancer. However, the interpretation of the efficacy data is complex, as in many cases the concurrent administration with chemotherapy led to excessive toxicity, compromising adequate delivery of dose intensity, which is a key factor for chemotherapy-derived benefit (Budman, 2004; Baselga et al, 2012; Bergh et al, 2012). Novel molecules such as nintedanib show a better preclinical profile and thus could overcome such limitation (Boehringer Ingelheim, GmbH, 2009). This feature would make it a potential good therapeutic option, given the activity of antiangiogenics in this malignancy.
We present here a phase I trial of the combination of nintedanib with standard treatment for neoadjuvant breast cancer. The RP2D is 150 mg per day BID combined with weekly paclitaxel (80 mg m2), followed by AC. At this dose level, the observed toxicity is virtually indistinguishable from that reported for weekly paclitaxel alone. Only minor, reversible (without treatment discontinuation) transaminases elevation was noticed. Indeed, the dose intensity of both drugs was close to 100%. Interestingly, class-specific effects such as hypertension of hand–foot syndrome were not observed for nintedanib at the RP2D. The efficacy at this dose level, in a cohort of patients with locally advanced breast cancer and mostly node positive, is very promising despite the low number of patients, achieving conservative surgery in most cases, and an unusually high rate of pathologic complete response. In light of these results, a randomised phase II trial in neoadjuvant HER-2-negative breast cancer with a target enrolment of 130 patients is ongoing. Of note, in this study, the response was measured following the Miller and Payne system as the response rate was not a primary end point; however, the randomised trial efficacy will be assessed based on the updated Symmans and Pusztai criteria, which may reflect more accurately the true activity of the combination (Symmans et al, 2007).
Change history
09 September 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Baselga J, Segalla JG, Roche H, Del Giglio A, Pinczowski H, Ciruelos EM, Filho SC, Gomez P, Van Eyll B, Bermejo B, Llombart A, Garicochea B, Duran MA, Hoff PM, Espie M, de Moraes AA, Ribeiro RA, Mathias C, Gil Gil M, Ojeda B, Morales J, Kwon Ro S, Li S, Costa F (2012) Sorafenib in combination with capecitabine: an oral regimen for patients with HER2-negative locally advanced or metastatic breast cancer. J Clin Oncol 30: 1484–1491.
Bear HD, Tang G, Rastogi P, Geyer CE Jr, Robidoux A, Atkins JN, Baez-Diaz L, Brufsky AM, Mehta RS, Fehrenbacher L, Young JA, Senecal FM, Gaur R, Margolese RG, Adams PT, Gross HM, Costantino JP, Swain SM, Mamounas EP, Wolmark N (2012) Bevacizumab added to neoadjuvant chemotherapy for breast cancer. N Engl J Med 366: 310–320.
Bergh J, Bondarenko IM, Lichinitser MR, Liljegren A, Greil R, Voytko NL, Makhson AN, Cortes J, Lortholary A, Bischoff J, Chan A, Delaloge S, Huang X, Kern KA, Giorgetti C (2012) First-line treatment of advanced breast cancer with sunitinib in combination with docetaxel versus docetaxel alone: results of a prospective, randomized phase III study. J Clin Oncol 30: 921–929.
Boehringer Ingelheim GmbH (2009) BIBF1120 Investigators Brochure. BI GmbH: Ingelheim am Rhein, Germany.
Budman DR (2004) Dose and schedule as determinants of outcomes in chemotherapy for breast cancer. Semin Oncol 31: 3–9.
Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Graf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, Langerod A, Green A, Provenzano E, Wishart G, Pinder S, Watson P, Markowetz F, Murphy L, Ellis I, Purushotham A, Borresen-Dale AL, Brenton JD, Tavare S, Caldas C, Aparicio S (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486: 346–352.
Davies C, Pan H, Godwin J, Gray R, Arriagada R, Raina V, Abraham M, Medeiros Alencar VH, Badran A, Bonfill X, Bradbury J, Clarke M, Collins R, Davis SR, Delmestri A, Forbes JF, Haddad P, Hou MF, Inbar M, Khaled H, Kielanowska J, Kwan WH, Mathew BS, Mittra I, Muller B, Nicolucci A, Peralta O, Pernas F, Petruzelka L, Pienkowski T, Radhika R, Rajan B, Rubach MT, Tort S, Urrutia G, Valentini M, Wang Y, Peto R (2013) Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 381: 805–816.
Earl HM, Vallier AL, Hiller L, Fenwick N, Young J, Iddawela M, Abraham J, Hughes-Davies L, Gounaris I, McAdam K, Houston S, Hickish T, Skene A, Chan S, Dean S, Ritchie D, Laing R, Harries M, Gallagher C, Wishart G, Dunn J, Provenzano E, Caldas C (2014) Effects of the addition of gemcitabine, and paclitaxel-first sequencing, in neoadjuvant sequential epirubicin, cyclophosphamide, and paclitaxel for women with high-risk early breast cancer (Neo-tAnGo): an open-label, 2 × 2 factorial randomised phase 3 trial. Lancet Oncol 15: 201–212.
Foulkes WD, Smith IE, Reis-Filho JS (2010) Triple-negative breast cancer. N Engl J Med 363: 1938–1948.
Ledermann JA, Hackshaw A, Kaye S, Jayson G, Gabra H, McNeish I, Earl H, Perren T, Gore M, Persic M, Adams M, James L, Temple G, Merger M, Rustin G (2011) Randomized phase II placebo-controlled trial of maintenance therapy using the oral triple angiokinase inhibitor BIBF 1120 after chemotherapy for relapsed ovarian cancer. J Clin Oncol 29: 3798–3804.
Mross K, Stefanic M, Gmehling D, Frost A, Baas F, Unger C, Strecker R, Henning J, Gaschler-Markefski B, Stopfer P, de Rossi L, Kaiser R (2010) Phase I study of the angiogenesis inhibitor BIBF 1120 in patients with advanced solid tumors. Clin Cancer Res 16: 311–319.
NCI National Cancer Institute: Common Terminology Criteria for Adverse Events, Version 4.0 http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm.
Ogston KN, Miller ID, Payne S, Hutcheon AW, Sarkar TK, Smith I, Schofield A, Heys SD (2003) A new histological grading system to assess response of breast cancers to primary chemotherapy: prognostic significance and survival. Breast 12: 320–327.
Reck M, Kaiser R, Mellemgaard A, Douillard JY, Orlov S, Krzakowski M, von Pawel J, Gottfried M, Bondarenko I, Liao M, Gann CN, Barrueco J, Gaschler-Markefski B, Novello S (2014) Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomised controlled trial. Lancet Oncol 15: 143–155.
Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, Burleigh A, Yap D, Bernard V, McPherson A, Shumansky K, Crisan A, Giuliany R, Heravi-Moussavi A, Rosner J, Lai D, Birol I, Varhol R, Tam A, Dhalla N, Zeng T, Ma K, Chan SK, Griffith M, Moradian A, Cheng SW, Morin GB, Watson P, Gelmon K, Chia S, Chin SF, Curtis C, Rueda OM, Pharoah PD, Damaraju S, Mackey J, Hoon K, Harkins T, Tadigotla V, Sigaroudinia M, Gascard P, Tlsty T, Costello JF, Meyer IM, Eaves CJ, Wasserman WW, Jones S, Huntsman D, Hirst M, Caldas C, Marra MA, Aparicio S (2012) The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486: 395–399.
Swain SM, Tang G, Geyer CE Jr., Rastogi P, Atkins JN, Donnellan PP, Fehrenbacher L, Azar CA, Robidoux A, Polikoff JA, Brufsky AM, Biggs DD, Levine EA, Zapas JL, Provencher L, Northfelt DW, Paik S, Costantino JP, Mamounas EP, Wolmark N (2013) Definitive results of a phase III adjuvant trial comparing three chemotherapy regimens in women with operable, node-positive breast cancer: the NSABP B-38 trial. J Clin Oncol 31: 3197–3204.
Symmans WF, Peintinger F, Hatzis C, Rajan R, Kuerer H, Valero V, Assad L, Poniecka A, Hennessy B, Green M, Buzdar AU, Singletary SE, Hortobagyi GN, Pusztai L (2007) Measurement of residual breast cancer burden to predict survival after neoadjuvant chemotherapy. J Clin Oncol 25: 4414–4422.
von Minckwitz G, Eidtmann H, Rezai M, Fasching PA, Tesch H, Eggemann H, Schrader I, Kittel K, Hanusch C, Kreienberg R, Solbach C, Gerber B, Jackisch C, Kunz G, Blohmer JU, Huober J, Hauschild M, Fehm T, Muller BM, Denkert C, Loibl S, Nekljudova V, Untch M (2012) Neoadjuvant chemotherapy and bevacizumab for HER2-negative breast cancer. N Engl J Med 366: 299–309.
Weis SM, Cheresh DA (2011) Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med 17: 1359–1370.
Acknowledgements
This work was funded by grants FIS PI10/0288 and FIS PI13/00430 and donations from Rosae Foundation and Avon España S.A.U. M.Q.-F. was a recipient of a 2010 Beca-Retorno from the AECC Scientific Foundation. Boehringer-Ingelheim provided financial support for this study. In addition, nintedanib was supplied by Boehringer-Ingelheim.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Quintela-Fandino, M., Urruticoechea, A., Guerra, J. et al. Phase I clinical trial of nintedanib plus paclitaxel in early HER-2-negative breast cancer (CNIO-BR-01-2010/GEICAM-2010-10 study). Br J Cancer 111, 1060–1064 (2014). https://doi.org/10.1038/bjc.2014.397
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.397
Keywords
This article is cited by
-
Synthesis and evaluation of in vitro cytotoxic effects of triazol/spiroindolinequinazolinedione, triazol/indolin-3-thiosemicarbazone and triazol/thiazol-indolin-2-one conjugates
DARU Journal of Pharmaceutical Sciences (2020)
-
Therapeutic impact of Nintedanib with paclitaxel and/or a PD-L1 antibody in preclinical models of orthotopic primary or metastatic triple negative breast cancer
Journal of Experimental & Clinical Cancer Research (2019)
-
Nintedanib plus letrozole in early breast cancer: a phase 0/I pharmacodynamic, pharmacokinetic, and safety clinical trial of combined FGFR1 and aromatase inhibition
Breast Cancer Research (2019)
-
Variable impact of three different antiangiogenic drugs alone or in combination with chemotherapy on multiple bone marrow-derived cell populations involved in angiogenesis and immunity
Angiogenesis (2019)
-
The tyrosine kinase inhibitor nintedanib activates SHP-1 and induces apoptosis in triple-negative breast cancer cells
Experimental & Molecular Medicine (2017)
