
Abstract
Background:
Tamoxifen has anti-oestrogenic and anti-tumour activity in the breast, but is oestrogenic and carcinogenic in the endometrium. It can induce experimental tumours by both hormonal and DNA-damaging mechanisms, but its carcinogenic mode of action in human endometrium remains unclear.
Methods:
We investigated whether an epigenetic mechanism, involving promoter hypermethylation of the gene for the DNA repair enzyme MGMT (O6-methylguanine DNA methyltransferase), was associated with K-RAS, TP53 and PTEN mutations in endometrial tumours from women treated with tamoxifen (TAM, n=30) or unexposed to the drug (EC, n=38).
Results:
There were significant (P<0.05) differences in tumour grade between the TAM and EC groups, with more favourable morphology in the latter. K-RAS mutations, predominantly G>A, occurred in small numbers in both groups. TP53 mutations were of mainly A>G, C>T and indel modifications in both groups, but more frequent in TAM cases. PTEN mutations dominated in EC tumours and were of the type that has large impact on protein function, such as indel or nonsense mutations. These observations alongside the mutational spectrum in PTEN suggest that the malignancies arise from different backgrounds, hence pointing to an effect of tamoxifen. Both groups displayed MGMT promoter hypermethylation. This coincided with mutations more frequently in the TAM (78%) than in the EC (50%) group, even though there were significantly (P<0.05) fewer mutations and methylations in TAM cases.
Conclusions:
Although the difference in coincidence did not reach significance with the current sample size, the findings suggest that epigenetic processes may play a role in the way tamoxifen induces endometrial cancer.
Similar content being viewed by others

Main
Endometrial cancer is the fourth most common cancer in women in industrialised countries, with many risk factors including the use of exogenous oestrogens and certain drugs, such as tamoxifen (Fisher et al, 1998). Tamoxifen is a selective oestrogen receptor modulator, which acts antagonistically in oestrogen-dependent breast cancer by competing for the oestrogen receptor (ER), thereby forming a nuclear complex that recruits co-repressors to stop oestrogen-driven transcription. This decreases DNA synthesis and causes G0/G1 phase arrest of the cell cycle (Shang et al, 2000). In human endometrium, however, tamoxifen displays oestrogenic activity.
Tamoxifen gives rise to DNA adducts and liver cancer in both male and female rats (Davis et al, 2000). In rats, the minor metabolite alpha-OH-tamoxifen is a substrate for hydroxysteroid sulphotransferase a, which leads to bioactivation in the liver, whereas in humans the major metabolite 4-OH-tamoxifen is conjugated by oestrogen sulphotransferases leading to detoxification (Chen et al, 2002). This interspecies difference in tamoxifen metabolism may partly explain why the compound is more toxic for rats compared to humans. Nonetheless, the potential neoplastic effects of tamoxifen in human endometrium have been largely unexplored at the molecular level.
In humans, a major circulating inactive form of plasma oestrogen is oestrogen sulphate (E1S and E2S) that can be activated by intra-tumoural steroid sulphatases, thereby exerting additional in situ hormonal activity in oestrogen-dependent breast cancers (Chetrite et al, 2000). Similar observations have been made in endometrial tumours displaying increased steroid sulphatase and lowered oestrogen sulphotransferase activity (Utsunomiya et al, 2004), and in endometrial cancer cells where tamoxifen was shown to perturb the balance of oestrogen-metabolising enzymes and disposition of oestrogen metabolites (Williams-Brown et al, 2011).
Metabolism of tamoxifen also involves formation of quinone metabolites, which can oxidise and alkylate cellular macromolecules in vivo (Zhang et al, 2000). O6-methylguanine DNA methyltransferase (MGMT) is responsible for repairing alkylation DNA damage and also has a possible role in inhibiting ER-mediated cell proliferation (Teo et al, 2001). In addition, certain polymorphic variants of MGMT may influence endometrial cancer risk (Han et al, 2006). Expression of MGMT is in part controlled through epigenetic processes such as hypermethylation of the promoter region of the gene, which can lead to inactivation of the repair pathway (Esteller et al, 1999), but evidence for the role of MGMT in endometrial cancer is conflicting (Furlan et al, 2006; Rimel et al, 2009).
K-RAS, PTEN and TP53 are often mutated in endometrial cancer, at frequencies that vary with tumour grade (Liu, 2007; Bansal et al, 2009). The aim of this project was to investigate associations between tamoxifen exposure, MGMT promoter hypermethylation and mutations in TP53, PTEN and K-RAS in endometrial tumours and tissue, in order to shed light on the carcinogenic mode of action of tamoxifen in humans.
Materials and Methods
Patients
All samples were from cancer cases and subdivided according to the following criteria:
TAM (n=30): endometrial tumours from women who had taken tamoxifen; average age 69.4±11.2 years; 10 with matching blood samples.
EC (n=38): endometrial tumours from patients not treated with tamoxifen; average age 67.0±9.4 years; 24 with matching blood samples.
Treatment with tamoxifen ranged from 4 months to 20 years at 20 mg per day. In the majority of cases the tamoxifen use was restricted to approximately 60 months duration. Extended usage of tamoxifen was only in two cases with breast cancer recurrences.
The samples were from patients undergoing gynaecological surgery in the Royal Preston Hospital of Lancashire Teaching Hospitals NHS Foundation Trust in Preston between January 2006 and August 2012. Ethical committee approvals were obtained (Local Research Ethical Committee (LREC) approval no. 05/Q1302/83 and the Research and Ethics Committee (REC) approval no. 10/H0308/75). The consenting patients were between the ages of 37 and 90 years. Samples were formalin-fixed and paraffin-embedded (FFPE) and/or flash frozen (FF). Histopathology was performed to determine the tumour type and grade. Shavings of the FFPE blocks and/or small pieces of the FF tissues, as well as frozen blood, were sent to King’s College London for DNA isolation.
One patient in each group was found to have a silent germline mutation in TP53. These patients were excluded from the data compilation (Figures and Tables) and statistical calculation, although still included in Supplementary data.
Methylation specific-PCR analysis
DNA was extracted from triplicate shavings (10-μm thickness) from tumour samples containing less than 10% normal tissue using a QIAamp DNA FFPE Tissue Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s instructions. The extracted DNA was measured spectrophotometrically for purity and concentration, and 1 μg (where possible) was bisulphite-treated using the Epitect Bisulfite Kit (Qiagen GmbH, Hilden, Germany). The bisulphite-converted DNA was amplified by PCR for the selected promoter region with the following primers synthesised in 25 nmol scale by Invitrogen (Carlsbad, CA, USA):
MGMT-MF: TTTCGACGTTCGTAGGTTTTCGC
MGMT-MR: GCACTCTTCCGAAAACGAAACG
MGMT-UF: TTTGTGTTTTGATGTTTGTAGGTTTTTGT
MGMT-UR: AACTCCACACTCTTCCAAAAACAAAACA
Primers were diluted in distilled water to 300 ng μl−1 and the following PCR mixture was prepared; 5 μl of 10 × PCR reaction buffer, 5 μl of 50 mM MgCl2 and 0.2 μl Platinum Taq (1U; all from Invitrogen) was mixed with 5 μl 10 mM dNTP-mix (Qiagen), 2 × 1 μl primer (forward and reverse), 10 μl DNA template and 22.8 μl of distilled water to a total volume of 50 μl. Bisulphite-converted methylated and unmethylated human DNA, as well as non-converted unmethylated DNA that was converted along with the samples, and pure water, were included with each PCR analysis as positive or negative controls.
The thermocycler was programmed for a 5-min hot start at 95 °C, followed by 40 cycles of 94 °C for 40 s, 59 °C for 40 s and 72 °C for 40 s, and finally a 10-min elongation step at 72 °C. The PCR products were separated on 3% agarose at 140 V and visualised with EtBr. The bisulphite treatment and PCR amplification were carried out in triplicate; if a methylated band was observed at least two out of three times for a given sample, then it was deemed to be positive for methylation.
Sequencing
DNA was isolated from FFPE tissues as described above, and from FF tissues and blood using a DNeasy Blood & Tissue kit, (Qiagen). The purified DNA was measured for concentration and aliquots were portioned into 96-well plates and immediately frozen at −80 °C. Sequencing was performed by Beckman Coulter Genomics (United States) on K-RAS exon 1–4, PTEN exons 1–9 and TP53 exons 4–11.
Statistical analysis
Statistical analysis was performed using a two-tailed Z-test at the 95% confidence level by evaluating the proportions of given occurrences in the different patient groups (TAM and EC). Differences were statistically significant when P⩽0.05.
Results
Tumour histology and grading
Patients with tamoxifen-associated endometrial cancer (TAM) had significantly lower occurrence (P⩽0.05) of type I tumours than those in the non-exposed (EC) group with 5 out of 29 (17%) and 17 out of 37 (46%), respectively (Table 1). TAM cases had more type II tumours of predominantly serous, carcinosarcoma and mixed carcinoma, whereas in the EC group these were mainly grade 3 endometrioid carcinomas followed by clear cell and carcinosarcoma (Table 1). Grouping together the endometrioid tumours of all grades (G1–G3), a significant (P⩽0.05) overrepresentation of 23 out of 27 (62%) was found among the EC cases. There was also a significant difference (P⩽0.05) between these two groups with respect to other diverse lesions, including hyperplasia, polyps, inactive, menstrual, proliferative and secretory endometrium, which were completely absent from the EC group, but represented about 4 out of 29 (14%) of the malignancies among the TAM cases.
MGMT promoter hypermethylation
Patients scored positive for promoter hypermethylation in both groups (Table 2), with TAM 9 out of 29 (31%) and EC 14 out of 37 (38%). There was no statistical difference between any of the groups and no correlation between the type or grade of tumours and MGMT promoter hypermethylation (Supplementary data).
Mutation analysis
Overall mutations
Counting the number of patients with mutations, regardless of type of gene or multiple mutations in the same gene, revealed no difference between the two tumour groups. There was a trend in higher occurrence of methylations or mutations in the EC as compared to the TAM group, which was significant (P⩽0.05) when adding up patients with either of the modifications present. The reverse was observed in patients who exhibited both a mutation and methylation (Table 2). Thus, although not statistically verified at the 95th percentile, a closer look at how many patients with methylation also exhibited mutations showed that this occurrence was higher in the TAM compared with the EC group, with 7 of 9 (78%) and 7 of 14 (50%), respectively (Table 2).
Specific mutations
K-RAS mutations were found in both groups and were mainly of the missense type and distributed over codons 11–13 in exon 2; four in TAM and two in EC (Table 3). The mutations in the TAM group consisted of one G>T transversion and three G>A transitions. In the EC group, the K-RAS mutations were one G>A transition at codon 13 and a T>C transversion (Supplementary data).
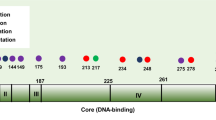
TP53 mutations occurred in both groups to a similar extent (34% in TAM and 38% in EC) and were of diverse types (Table 3). The tumour groups displayed mainly missense or indel alterations, with the addition of a few nonsense mutations in EC, and predominantly clustering within the central DNA-binding core domain within residues 102–292. Some of the missense mutations designated with SNP ascension numbers were found to be associated to Li-Fraumeni syndrome (see Discussion). Two patients, one in each group, had a germline silent mutation in TP53 (Arg213Arg rs1800372), which was confirmed in corresponding blood samples (Supplementary data).
PTEN was extensively mutated in the EC group (Table 3) and found within varying grades and types of tumours and of the sort that has great impact on protein function; i.e., truncated via a stop-codon, or frameshift through either insertion or deletion. In fact, out of the 22 PTEN mutations observed in the EC group, 11 were either stop or indel (supplementary data). Only six patients from the TAM group displayed PTEN mutations, out of which five were indel and just one was a silent mutation (Table 3). Most alterations occurred in exon 5 (photsphatase domain and core motif), 7–8 and one mutation each in exons 3, 4, 6 and 9 (C-terminal region). No correlation to methylation status was found in any of the cases.
The mutation spectrum of the two tumour groups showed that the most commonly mutated gene following tamoxifen treatment was TP53, with A>G (27%), C>T (20%) and indel (30%) modifications (Figure 2). In the PTEN gene, indel was by far the most common alteration with 83%. The EC group displayed high degree of mutation in both TP53 and PTEN. In TP53, the most prominent base-change was C>T (43%), followed by A>G (22%) and indel (21%), and occurred mainly in tumours of non-endometrioid character. In PTEN, C>T (18%) and indel (23%) dominated, and over half of all mutations were found in endometrioid tumours (Supplementary data).
Interpretation
Epidemiological studies have shown an increased risk of endometrial cancer with tamoxifen exposure, correlating risks ranging from 2.53 to 7.5 with length of exposure/treatment (van Leeuwen et al, 1994; Fisher et al, 1998). The mechanisms of action are yet to be fully evaluated; despite the demonstration of the DNA adduct forming capacity of metabolically activated tamoxifen in animals and in some experimental systems, there is conflicting evidence on the genotoxicity of tamoxifen in humans (Phillips, 2001; Kim et al, 2004; Singh et al, 2007, 2008). Here we aimed to investigate if there was a possible connection between tamoxifen treatment, promoter hypermethylation of MGMT and mutations in genes known to play a role in endometrial cancer.
Tumour histology and grading
High-grade malignancies were more frequent in the TAM group compared to EC, which consisted mainly of type I category malignancies of endometrioid character and G1–G2 morphology (Table 1), corroborating previous observations that women who underwent tamoxifen treatment for breast cancer and subsequently developed uterine cancers had higher-grade tumours with less favourable prognosis (Magriples et al, 1993; van Leeuwen et al, 1994). A possible bias towards high-grade tumours at medical centres that are better equipped for treating more severe cases is disputed based on the scarcity of the tamoxifen-exposed cases even with influx of large patient numbers, the endeavours of peripheral centres to report and collect all available samples, and that none of the patients opted out of the study despite the length of time. In addition, the medical histories of the patients were taken into consideration, where previous/adjuvant treatments in patients with breast cancer in the TAM group involved either radiotherapy or a combination of 5FU/epirubicin/cyclophosphamide. These therapies and this combination of drugs have not been reported in the literature to be associated with increased risk of endometrial cancer.
When assessing the occurrence of type I tumours, there was a statistically significant difference (P⩽0.05) between the TAM and EC groups, which also held true when evaluating the occurrence of all endometrioid morphologies as a whole (G1–G3; Table 1). It is reported that the majority of endometrial cancers (70–80%), designated as type I carcinomas, follow the oestrogen-related pathway and seemingly arise in the background of hyperplastic endometrium with endometrioid differentiation (Liu, 2007). However, many endometrial carcinomas are overlapping in their clinical, morphologic, immunohistochemical and molecular features between types I and II. Lesions with various histologies including polyps, hyperplasia, late/mid-secretory and proliferative endometrium, and progesterone effect were only observed in the TAM group. Many of these are normal features and are universal components of the menstrual cycle, although some forms of hyperplasia (especially complex atypical) can be premalignant. Polyps are, for instance, not precancerous, but still occur more commonly in women on tamoxifen (Biron-Shental et al, 2003).
MGMT promoter hypermethylation
There was no statistical difference between groups with regard to MGMT promoter hypermethylation, which was seen in 9 out of 29 (31%) and 14 out of 37 (38%) cases, in TAM and EC groups, respectively (Table 2). There is some support for this in the literature, but also some studies that contradict our finding. Rimel et al (2009) found an absence of MGMT promoter hypermethylation in 120 endometrial cancer cases, which was also supported by previous findings in a small number (n=17) of endometrial cancers (Jacinto and Esteller, 2007). On the other hand, in a study involving synchronous ovarian and endometrial primary tumours there was a similar rate of MGMT promoter hypermethylation to what we report here (Furlan et al, 2006). One aspect to consider is methodology where it can be argued that studies finding positive methylation were done with MS-PCR, which may increase sensitivity and methylation-positive cases with increasing cycles of amplification (Shen et al, 2005). However, different methodologies (MS-PCR and/or combined bisulphite restriction analysis) have been used on various DNA sources, often in parallel, and results were found to be consistent (Xiong and Laird, 1997; Shen et al, 2005; Cheng et al, 2010). In addition, primer design needs to meet certain criteria (Herman et al, 1996; Brandes et al, 2007) and proper controls need to be added in the analysis to avoid problems of false-positives. Hence, we optimised the melting temperature with gradient PCR, using both converted and non-converted DNA and water to minimise false-positives and these controls were included in every amplification step from then on (Figure 1B). The quality of the DNA does not explain discordance with the literature, as methylation was also observed in both groups and no difference in the extent of methylation was seen between FFPE or flash-frozen tissues. When comparing methylation to tumour type there were no significant correlations.

Examples of MS-PCR analysis (A; showing the first lane of gels, including 10 patient samples of each group). ‘M’ represents methylated bands and ‘U’ unmethylated. Controls (B) were used at each analysis with BC-U being unmethylated human DNA that was bisulphite converted during sample workup procedures to assess the success of the conversion step. ‘C–U’ and ‘C–M’ were commercially available bisulphite converted unmethylated and methylated human DNA, respectively. ‘DNA’ represents non-converted human DNA. The H2O samples were lacking DNA of any sort in the PCR mixture. Note that close to the bottom line of the ladder corresponding to about 50 bp, bands can be seen. These are primer dimers. The product bands for both ‘M’ and ‘U’ are around 100 bp with only minor differences.
Mutation analysis
K-RAS mutations are fairly common (10–30%) in endometrial cancer, and are mostly found in lower-grade tumours (Liu, 2007). Tamoxifen gave rise to G>T transversions at codon 12 in exon 2 in the K-RAS gene in rat liver (Davies et al, 1999), whereas in human endometrium these mutations are less frequent than G>A transitions (Hachisuga et al, 2005). In our study, the only type of K-RAS mutation, which has been associated to adverse outcome, was G>A transition resulting in missense mutations (Gly>Ser or Gly>Asp). This type of mutation at codon 12 has been observed in endometrial cancer following tamoxifen treatment (Prasad et al, 2005; Wallen et al, 2005), but is also common in colon (Brink et al, 2003; Wu et al, 2005) and pancreatic cancer (Hruban et al, 1993). Some studies show a possible association between MGMT promoter hypermethylation and K-RAS G>A transition with regard to colon cancer (Shen et al, 2005; Nagasaka et al, 2008). Even if the rate of K-RAS mutations in our study falls within the reported values (5–14%, Table 3), no conclusions can be drawn with regard to mutation type and methylation. One patient from the EC group displayed a T>C transition at codon 173 (rs17473423), but it is a common polymorphism and no convincing data have been found associating it with any adverse outcome, although it was seen in a few cases of Noonan Syndrome, which is a relatively common autosomal dominant congenital disorder (Yamamoto et al, 2007).
In a study by Turbiner et al (2008), K-RAS mutations appeared to be more common among endometrial carcinomas in patients with history of tamoxifen exposure (21%) compared with informative endometrial carcinomas (11%), and were comparably low in both tumour-adjacent groups, although the difference was not statistically significant. Previous reports also found that endometrial polyps in patients treated with tamoxifen had a significantly higher frequency of K-RAS mutations compared with sporadic endometrial hyperplasia (Hachisuga et al, 2003) and treatment with tamoxifen or toremifene gave rise to de novo K-RAS mutation in a seemingly normal endometrial tissue (Wallen et al, 2005). Accordingly, our study showed K-RAS mutations in 4 out of 20 (20%) mutations among TAM patients, and 2 out of 38 (5%) in the EC cases.
PTEN mutations or sequence alterations were more prominent in the EC group with endometrioid tumours overrepresented (Tables 1 and 3) and predominantly of the sort that has great impact on protein function, such as nonsense mutations or frameshifts, and clustering mainly at exon 5, with two that fell within the phosphatase catalytic domain at codons 122–132, corroborating published observations (Prasad et al, 2005; Bansal et al, 2009). Residues 233–240 and 308–315 seem to be potential tyrosine phosphate acceptor sites (Steck et al, 1997), and thus the nonsense mutation at codon 233 (rs121909219) is associated with both Bannayan–Zonnana syndrome and Cowden’s disease (Ali et al, 1999). In addition, several mutations flank closely these crucial sites, although not within the regions. Some of these were Arg130Gly rs121909224 and Asp252Val rs121909239, and were all disease associated in numerous malignancies, among them endometrial cancer (Cohen et al, 2010).
Only six patients with PTEN alterations were from the TAM group with five indel and a silent mutation (Table 3). The difference in the number of patients with PTEN mutation between the TAM and EC groups was statistically significant with P⩽0.05 at the 95% confidence level. This is in agreement with a similar observation in the literature, where PTEN mutations occurred less frequently in the tamoxifen-treated patients, although not statistically significant due to that cohort size (Turbiner et al, 2008).
According to the literature TP53 mutations in endometrial cancer are more abundant in high-grade tumours with poor prognosis that are often oestrogen independent (Lax et al, 2000; Bansal et al, 2009). In our study, most of the TP53 mutations occur in the serous, clear-cell or serous/clear-cell tumours in the TAM group. In exon 7, two missense mutations (Arg273His rs28934576 and Arg282Trp rs28934574) were found associated with Li-Fraumeni syndrome (Malkin et al, 1992; Toguchida et al, 1992). Only three tumours were of other morphology; i.e., adenocarcinoma, carcinosarcoma and sarcoma with high-grade FIGO 1B. In the EC group, a similar pattern emerged, where the majority of TP53 mutations clustered in serous, clear-cell or serous/clear-cell tumours and only three were in either carcinosarcoma or endometrioid tumours. In the EC group 38% of the mutations were found in TP53, with a mix of missense, homoindel and stop mutations. In exon 6 a missense mutation was found with SNP accession number rs28934575, also present in Li-Fraumeni syndrome (Toguchida et al, 1992).
It has been reported that TP53 mutations do not concur with either PTEN (Koul et al, 2002) or K-RAS (Lax et al, 2000) mutations in endometrial cancer, but rather with loss of p16. This is in part what has been observed in this study, as virtually none of the mutations in either of the genes coincided. However, it has been shown that TP53 gene mutations occur in some endometrioid endometrial cancers in the presence of PTEN mutations, suggesting that both these genes participate in the development of these tumours (Janiec-Jankowska et al, 2010).
Our study supports in part some observations made by Prasad et al (2005), that endometrial tumours from these two groups contain similar genetic alterations with regard to the type of gene mutations. However, the mutational spectrum within some of the genes is clearly different (Figure 2). Although not much can be said for K-RAS due to the low occurrence, and the similarities in TP53, PTEN mutations are quite different between tamoxifen-exposed and non-exposed cases, suggesting the malignancies arise from different backgrounds, hence pointing to an effect of tamoxifen.

Mutational spectrum of K-RAS, TP53 and PTEN in tumour tissues from TAM and EC patients.
We have also observed an interesting coincidence between methylation and mutation, which was regardless of tumour morphology or genes affected (Table 2). This coincidence was between 7 of 9 (78%) and 7 of 14 (50%) in TAM and EC, respectively. The higher occurrence of this in TAM, despite the significantly lower levels of either alterations in this group, suggests that there may be an effect of tamoxifen exposure, although a larger number of cases would be needed to assess this fully. This suggests that tamoxifen exposure, apart from hormonal effects, possible adduct forming capacity, may also be driving malignancy through epigenetic processes.
Conclusion
Patients receiving tamoxifen developed more high-grade tumours with less favourable prognosis. Genes commonly mutated were TP53 and PTEN, with TP53 being the predominant one in the TAM cases and PTEN in the EC group. We also confirmed the presence of MGMT promoter hypermethylation and, in addition, a coincidence between methylation and mutation in both groups. The occurrence of this, however, was higher in the TAM group, although not significantly. Our findings suggest that hypermethylation of MGMT promoter may play an epigenetic role in the induction of endometrial cancer by tamoxifen.
Change history
10 June 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ali IU, Schriml LM, Dean M (1999) Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. J Natl Cancer Inst 91: 1922–1932.
Bansal N, Yendluri V, Wenham RM (2009) The molecular biology of endometrial cancers and the implications for pathogenesis, classification, and targeted therapies. Cancer Control 16: 8–13.
Biron-Shental T, Tepper R, Fishman A, Shapira J, Cohen I (2003) Recurrent endometrial polyps in postmenopausal breast cancer patients on tamoxifen. Gynecol Oncol 90: 382–386.
Brandes JC, Carraway H, Herman JG (2007) Optimal primer design using the novel primer design program: MSPprimer provides accurate methylation analysis of the ATM promoter. Oncogene 26: 6229–6237.
Brink M, de Goeij AF, Weijenberg MP, Roemen GM, Lentjes MH, Pachen MM, Smits KM, de Bruine AP, Goldbohm RA, van den Brandt PA (2003) K-ras oncogene mutations in sporadic colorectal cancer in The Netherlands Cohort Study. Carcinogenesis 24: 703–710.
Chen G, Yin S, Maiti S, Shao X (2002) 4-Hydroxytamoxifen sulfation metabolism. J Biochem Mol Toxicol 16: 279–285.
Cheng ZD, Hu SL, Sun YB, Xu WP, Shen G, Kong XY (2010) Promoter methylation of CHFR gene in gastric carcinoma tissues detected using two methods. Chin J Cancer 29: 163–166.
Chetrite GS, Cortes-Prieto J, Philippe JC, Wright F, Pasqualini JR (2000) Comparison of estrogen concentrations, estrone sulfatase and aromatase activities in normal, and in cancerous, human breast tissues. J Steroid Biochem Mol Biol 72: 23–27.
Cohen Y, Shalmon B, Korach J, Barshack I, Fridman E, Rechavi G (2010) AKT1 pleckstrin homology domain E17K activating mutation in endometrial carcinoma. Gynecol Oncol 116: 88–91.
Davies R, Gant TW, Smith LL, Styles JA (1999) Tamoxifen induces G:C—>T:A mutations in the cII gene in the liver of lambda/lacI transgenic rats but not at 5′-CpG-3′ dinucleotide sequences as found in the lacI transgene. Carcinogenesis 20: 1351–1356.
Davis W, Hewer A, Rajkowski KM, Meinl W, Glatt H, Phillips DH (2000) Sex differences in the activation of tamoxifen to DNA binding species in rat liver in vivo and in rat hepatocytes in vitro: role of sulfotransferase induction. Cancer Res 60: 2887–2891.
Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG (1999) Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 59: 793–797.
Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N, Atkins J, Daly M, Wieand S, Tan-Chiu E, Ford L, Wolmark N (1998) Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst 90: 1371–1388.
Furlan D, Carnevali I, Marcomini B, Cerutti R, Dainese E, Capella C, Riva C (2006) The high frequency of de novo promoter methylation in synchronous primary endometrial and ovarian carcinomas. Clin Cancer Res 12: 3329–3336.
Hachisuga T, Miyakawa T, Tsujioka H, Horiuchi S, Emoto M, Kawarabayashi T (2003) K-ras mutation in tamoxifen-related endometrial polyps. Cancer 98: 1890–1897.
Hachisuga T, Tsujioka H, Horiuchi S, Udou T, Emoto M, Kawarabayashi T (2005) K-ras mutation in the endometrium of tamoxifen-treated breast cancer patients, with a comparison of tamoxifen and toremifene. Br J Cancer 92: 1098–1103.
Han J, Hankinson SE, De Vivo I (2006) Polymorphisms in O6-methylguanine DNA methyltransferase and endometrial cancer risk. Carcinogenesis 27: 2281–2285.
Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB (1996) Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 93: 9821–9826.
Hruban RH, van Mansfeld AD, Offerhaus GJ, van Weering DH, Allison DC, Goodman SN, Kensler TW, Bose KK, Cameron JL, Bos JL (1993) K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am J Pathol 143: 545–554.
Jacinto FV, Esteller M (2007) MGMT hypermethylation: a prognostic foe, a predictive friend. DNA Repair (Amst) 6: 1155–1160.
Janiec-Jankowska A, Konopka B, Goluda C, Najmola U (2010) TP53 mutations in endometrial cancers: relation to PTEN gene defects. Int J Gynecol Cancer 20: 196–202.
Kim SY, Suzuki N, Laxmi YR, Shibutani S (2004) Genotoxic mechanism of tamoxifen in developing endometrial cancer. Drug Metab Rev 36: 199–218.
Koul A, Willen R, Bendahl PO, Nilbert M, Borg A (2002) Distinct sets of gene alterations in endometrial carcinoma implicate alternate modes of tumorigenesis. Cancer 94: 2369–2379.
Lax SF, Kendall B, Tashiro H, Slebos RJ, Hedrick L (2000) The frequency of p53, K-ras mutations, and microsatellite instability differs in uterine endometrioid and serous carcinoma: evidence of distinct molecular genetic pathways. Cancer 88: 814–824.
Liu FS (2007) Molecular carcinogenesis of endometrial cancer. Taiwan J Obstet Gynecol 46: 26–32.
Magriples U, Naftolin F, Schwartz PE, Carcangiu ML (1993) High-grade endometrial carcinoma in tamoxifen-treated breast cancer patients. J Clin Oncol 11: 485–490.
Malkin D, Jolly KW, Barbier N, Look AT, Friend SH, Gebhardt MC, Andersen TI, Borresen AL, Li FP, Garber J, Strong LC (1992) Germline mutations of the p53 tumor-suppressor gene in children and young adults with second malignant neoplasms. N Engl J Med 326: 1309–1315.
Nagasaka T, Goel A, Notohara K, Takahata T, Sasamoto H, Uchida T, Nishida N, Tanaka N, Boland CR, Matsubara N (2008) Methylation pattern of the O6-methylguanine-DNA methyltransferase gene in colon during progressive colorectal tumorigenesis. Int J Cancer 122: 2429–2436.
Phillips DH (2001) Understanding the genotoxicity of tamoxifen? Carcinogenesis 22: 839–849.
Prasad M, Wang H, Douglas W, Barakat RR, Ellenson LH (2005) Molecular genetic characterization of tamoxifen-associated endometrial cancer. Gynecol Oncol 96: 25–31.
Rimel BJ, Huettner P, Powell MA, Mutch DG, Goodfellow PJ (2009) Absence of MGMT promoter methylation in endometrial cancer. Gynecol Oncol 112: 224–228.
Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103: 843–852.
Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR, Einspahr JG, Buckmeier J, Alberts DS, Hamilton SR, Issa JP (2005) MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst 97: 1330–1338.
Singh MN, Stringfellow HF, Paraskevaidis E, Martin-Hirsch PL, Martin FL (2007) Tamoxifen: important considerations of a multi-functional compound with organ-specific properties. Cancer Treat Rev 33: 91–100.
Singh MN, Stringfellow HF, Walsh MJ, Ashton KM, Paraskevaidis E, Abdo KR, Martin-Hirsch PL, Phillips DH, Martin FL (2008) Quantifiable mRNA transcripts for tamoxifen-metabolising enzymes in human endometrium. Toxicology 249: 85–90.
Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV (1997) Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 15: 356–362.
Teo AK, Oh HK, Ali RB, Li BF (2001) The modified human DNA repair enzyme O(6)-methylguanine-DNA methyltransferase is a negative regulator of estrogen receptor-mediated transcription upon alkylation DNA damage. Mol Cell Biol 21: 7105–7114.
Toguchida J, Yamaguchi T, Dayton SH, Beauchamp RL, Herrera GE, Ishizaki K, Yamamuro T, Meyers PA, Little JB, Sasaki MS, Ralph R, Weichselbaum RR, Yandell DW (1992) Prevalence and spectrum of germline mutations of the p53 gene among patients with sarcoma. N Engl J Med 326: 1301–1308.
Turbiner J, Moreno-Bueno G, Dahiya S, Sanchez-Estevez C, Hardisson D, Prat J, Oliva E, Palacios J (2008) Clinicopathological and molecular analysis of endometrial carcinoma associated with tamoxifen. Mod Pathol 21: 925–936.
Utsunomiya H, Ito K, Suzuki T, Kitamura T, Kaneko C, Nakata T, Niikura H, Okamura K, Yaegashi N, Sasano H (2004) Steroid sulfatase and estrogen sulfotransferase in human endometrial carcinoma. Clin Cancer Res 10: 5850–5856.
van Leeuwen FE, Benraadt J, Coebergh JW, Kiemeney LA, Gimbrere CH, Otter R, Schouten LJ, Damhuis RA, Bontenbal M, Diepenhorst FW, van Tinteren H, Coebergh JWW, Kiemeney LALM, Gimbrère CHF, Otter R, Schouten LJ, Damhuis RAM, Benraadtc J, Bontenbal M (1994) Risk of endometrial cancer after tamoxifen treatment of breast cancer. Lancet 343: 448–452.
Wallen M, Tomas E, Visakorpi T, Holli K, Maenpaa J (2005) Endometrial K-ras mutations in postmenopausal breast cancer patients treated with adjuvant tamoxifen or toremifene. Cancer Chemother Pharmacol 55: 343–346.
Williams-Brown MY, Salih SM, Xu X, Veenstra TD, Saeed M, Theiler SK, Diaz-Arrastia CR, Salama SA (2011) The effect of tamoxifen and raloxifene on estrogen metabolism and endometrial cancer risk. J Steroid Biochem Mol Biol 126: 78–86.
Wu CM, Tang R, Wang JY, Changchien CR, Hsieh LL (2005) Frequency and spectrum of K-RAS codons 12 and 13 mutations in colorectal adenocarcinomas from Taiwan. Cancer Genet Cytogenet 158: 55–60.
Xiong Z, Laird PW (1997) COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res 25: 2532–2534.
Yamamoto K., Yoshida R, Ogata T (2007) KRAS Analysis in 34 Noonan Syndrome Patients without PTPN11 Mutation. Clinical Pediatric Endocrinology 16: 99–101.
Zhang F, Fan PW, Liu X, Shen L, van Breeman RB, Bolton JL (2000) Synthesis and reactivity of a potential carcinogenic metabolite of tamoxifen: 3,4-dihydroxytamoxifen-o-quinone. Chem Res Toxicol 13: 53–62.
Acknowledgements
This work was supported by the Rosemere Cancer Foundation and Cancer Research UK. We thank the funding bodies for support throughout this project. Hospital and pathology staff who facilitated collection of the tissues for the purpose of the study are also thanked.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Nagy, E., Gajjar, K., Patel, I. et al. MGMT promoter hypermethylation and K-RAS, PTEN and TP53 mutations in tamoxifen-exposed and non-exposed endometrial cancer cases. Br J Cancer 110, 2874–2880 (2014). https://doi.org/10.1038/bjc.2014.263
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.263
Keywords
This article is cited by
-
Downregulation of MGMT promotes proliferation of intrahepatic cholangiocarcinoma by regulating p21
Clinical and Translational Oncology (2020)
-
A PTENtial cause for the selectivity of oncolytic viruses?
Nature Immunology (2016)
