
Abstract
Background:
We assessed the maximum tolerated regimen (MTR) and dose-limiting toxicities of pazopanib and lapatinib in combination with weekly paclitaxel, and the effect of pazopanib and lapatinib on paclitaxel pharmacokinetics.
Methods:
Patients received intravenous paclitaxel on days 1, 8, and 15 of a 28-day cycle concurrently with daily pazopanib and lapatinib. Dose levels of paclitaxel (mg m−2)/pazopanib(mg)/lapatinib(mg) were 50/400/1000, 50/800/1000, 80/800/1000, and 80/400/1000. At the MTR, additional patients were enrolled to further evaluate tolerability, and the potential effects of pazopanib and lapatinib, inhibitors of cytochrome P450 (CYP)3A4, on the pharmacokinetics of paclitaxel, a CYP2C8 and CYP3A4 substrate.
Results:
Twenty-six patients were enrolled. Dose-limiting toxicities at the MTR (80/400/1000) included grade 4 thrombosis and grade 3 aspartate aminotransferase elevation. Other toxicities included diarrhoea, neutropenia, fatigue, and liver enzyme elevations. Coadministration of pazopanib 400 mg and lapatinib 1000 mg increased paclitaxel maximum plasma concentration (38%) and area under the curve (37%) relative to paclitaxel alone. One patient with a salivary gland tumour had a partial response; three patients had stable disease (⩾6 months).
Conclusions:
Pazopanib 400 mg per day and lapatinib 1000 mg per day can be combined with paclitaxel 80 mg m−2 in 28-day cycles. Coadministration of pazopanib and lapatinib, weak inhibitors of CYP2C8 and CYP3A4, had an inhibitory effect on paclitaxel clearance.
Similar content being viewed by others

Main
Inhibition of both the vascular endothelial growth factor receptor and epidermal growth factor receptor (EGFR) signalling pathways in the treatment of solid tumours represents an important strategy in potentially overcoming resistance to single-agent therapies (Rubin and Duensing, 2006). Preclinical evidence shows that dual blockade of these receptors in several animal models for breast, lung, colon, and pancreatic cancer results in synergistic inhibition of tumour growth (Olaussen et al, 2009).
Pazopanib is an orally active, potent, selective small-molecule tyrosine kinase inhibitor of vascular endothelial growth factor receptor-1, -2, and -3, platelet-derived growth factor receptor-α and -β, and c-Kit (Harris et al, 2008). Pazopanib is approved for patients with metastatic renal cell carcinoma and advanced soft tissue sarcoma previously treated with chemotherapy (Sternberg et al, 2010; van der Graaf et al, 2012). Lapatinib is a reversible, dual inhibitor of human epidermal growth factor receptor type 2 (HER2) and EGFR, and has been shown to inhibit tumour growth and/or induce apoptosis in EGFR- and HER2-dependent tumour cell lines and xenografts (Konecny et al, 2006). Lapatinib is currently approved for patients with HER2-positive metastatic breast cancer in combination with capecitabine after prior therapy with anthracycline, taxane, and trastuzumab (Geyer et al, 2006). Lapatinib also is approved with letrozole in postmenopausal women with HER2-positive advanced breast cancer where endocrine therapy is indicated (Johnston et al, 2009). Paclitaxel is a microtubule inhibitor that has demonstrated activity in a variety of solid tumours. Paclitaxel given at low doses and frequent intervals has purported antiangiogenic effects and when combined with angiogenesis inhibitors, may enhance anticancer activity (Lau et al, 2004; Shaked et al, 2008). The addition of an angiogenesis inhibitor to paclitaxel-based therapy has demonstrated benefit in the treatment of several solid tumours, including non-small cell lung cancer (Sandler et al, 2006). We have previously shown that paclitaxel at 80 mg m−2 on days 1, 8, and 15 every 28 days can be safely combined with pazopanib at a daily dose of 800 mg (Tan et al, 2010).
Previous evaluation of the feasibility of combining pazopanib and lapatinib in a phase I study supports a recommended phase II dosing of pazopanib 800 mg and lapatinib 1500 mg daily (de Jonge et al, 2013). Investigators also reported an increase in the area under the plasma concentration–time curve (AUC) and maximum observed plasma concentration (Cmax) of pazopanib at these dose levels relative to administration of pazopanib alone, but not at pazopanib 400 mg and lapatinib 1000 mg. Clinical activity has been demonstrated with combined inhibition of the VEGF and EGF receptors in the treatment of several malignancies, including metastatic breast cancer. Lower doses of the combination were used in these phase II studies. A randomised phase II study of pazopanib 400 mg and lapatinib 1000 mg (n=77) compared with lapatinib 1500 mg (n=73) was conducted in patients with HER2-positive metastatic breast cancer as first-line therapy (Johnston et al, 2013). The combination improved the week-12 response rate to 36.2% compared with 22.2% for single-agent lapatinib in the modified intent-to-treat population. The activity of pazopanib and lapatinib has also been evaluated in a phase II trial in which patients with relapsed HER2-positive inflammatory breast cancer were randomised to pazopanib alone, lapatinib alone, or pazopanib and lapatinib. The overall response rates were 31%, 47%, and 58%, respectively, demonstrating that the pazopanib–lapatinib combination had improved activity compared with either single agent (Cristofanilli et al, 2013). In addition, the results of a phase II trial, AVANTHER, evaluated the combination of weekly paclitaxel and dual targeting of HER2 and VEGF with trastuzumab and bevacizumab as preoperative therapy in patients with HER2-positive Stage II or III breast cancer, followed by surgery and then adjuvant anthracycline and cyclophosphamide followed by trastuzumab. The pathologic complete-response rate in evaluable patients for whom surgical information was known was 44% (16 out of 36), which is indicative of favourable activity (Abad et al, 2012).
Given the preclinical and clinical data suggesting improved efficacy with coadministration of antiangiogenic agents and anti-EGFR agents due to cross-talk between vascular endothelial growth factor receptor and EGFR pathways, the safety of adding this combination to a commonly used chemotherapy backbone of weekly paclitaxel was explored. The objectives of this phase I trial were to determine the maximum tolerated regimen (MTR) and dose-limiting toxicities (DLTs) of pazopanib and lapatinib in combination with weekly paclitaxel, and to assess the effect of pazopanib and lapatinib on the pharmacokinetics of paclitaxel.
Patients and methods
Trial design
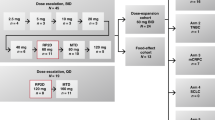
This was a phase I study (ClinicalTrials.gov identifier NCT00388076) to evaluate the safety, tolerability, and pharmacokinetic interactions of paclitaxel given on days 1, 8, and 15 of a 28-day cycle with pazopanib and lapatinib administered daily, conducted at three centres. The starting dose level of paclitaxel, pazopanib, and lapatinib was 50 mg m−2, 400 mg, and 1000 mg, respectively. These starting doses of pazopanib and lapatinib were chosen because a previous phase I study showed no drug–drug interaction between pazopanib and lapatinib at this level (de Jonge et al, 2013). Paclitaxel and pazopanib were escalated to a maximum of 80 mg m−2 and 800 mg per day, respectively, whereas lapatinib was maintained at 1000 mg per day for subsequent groups. At least three patients were enrolled at each dose level. If no DLTs were observed, escalation to the next dose level occurred. If one patient experienced a DLT, three additional patients were enrolled at that level. The dose of pazopanib or paclitaxel was escalated until the MTR was achieved. The MTR was defined as the highest dose at which no more than one in six patients experienced a DLT. Intrapatient dose escalation was not permitted. Once the MTR was established, up to nine additional patients were enrolled in an expansion phase to better evaluate the pharmacokinetics, safety, and tolerability of the combination at that dose level.
Adverse events were graded according to National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0. Dose-limiting toxicity was defined as any of the following events during cycle 1: grade 3 or 4 febrile neutropenia; grade 4 thrombocytopenia; grade 4 neutropenia more than 5 days; grade 4 hypertension or grade 3 hypertension that was not controlled with up to two antihypertensive medications; grade 4 proteinuria or grade 3 proteinuria persisting more than 3 weeks without improvement after discontinuation of pazopanib; inability to begin the next course of treatment within 2 weeks because of unresolved toxicity; any clinically significant grade 3 or 4 nonhematologic toxicity; or any grade 2 toxicity that was considered a DLT by the investigators.
Eligibility criteria
Eligible patients were 18 years of age or older with a histologic or cytologic diagnosis of cancer; an Eastern Cooperative Oncology Group performance status of 0 or 1; and adequate haematologic and organ function defined as an absolute neutrophil count ⩾2000 mm3, platelet count ⩾100 000 mm3, haemoglobin ⩾9 g dl−1, coagulation (prothrombin time, partial thromboplastin time, and international normalised ratio) ⩽1.2 × the upper limit of normal, calculated creatinine clearance >60 ml min−1, urine protein:creatinine ratio ⩽1, bilirubin ⩽1.5 × upper limit of normal, and aspartate and alanine aminotransferases (AST and ALT) ⩽2 × upper limit of normal. Major exclusion criteria included >3 prior lines of chemotherapy for metastatic disease, poorly controlled hypertension (systolic blood pressure ⩾140 mmHg or diastolic blood pressure ⩾90 mmHg), QTc interval ⩾470 msec, grade ⩾2 neuropathy, arterial or venous thrombi within 3 months or bleeding within 6 weeks of study entry, and any conditions affecting gastrointestinal absorption. Ethical approvals were obtained and the study protocol was approved by the institutional review board of each participating center. The trial was conducted in accordance with good clinical practice and the principles of the Declaration of Helsinki. All patients provided written informed consent before receiving treatment.
Treatment
Paclitaxel was commercially available and given intravenously as a 1-h infusion on days 1, 8, and 15 of a 28-day cycle after administration of pazopanib and lapatinib. Premedications for paclitaxel consisted of dexamethasone 10 mg or methylprednisolone 50 mg, diphenhydramine 50 mg, and ranitidine 50 mg, given per institutional policy. Pazopanib was supplied by GlaxoSmithKline (Research Triangle Park, NC, USA) as 200-mg and 400-mg capsule-shaped tablets. Lapatinib was also supplied by GlaxoSmithKline as 250-mg tablets. Both pazopanib and lapatinib were given orally once daily at least 1 h before or 2 h after a meal.
Assessments
At the beginning of each cycle, patients underwent a history and physical examination and laboratory assessments, including haematology, clinical chemistry, thyroid function tests, amylase, lipase, urine protein:creatinine ratio, and an electrocardiogram. On days 8 and 15 of each cycle, haematology and clinical chemistry were also assessed. Multigated acquisition scan or echocardiogram was obtained before treatment and before even-numbered cycles. Monitoring of thyroid function, amylase, and lipase occurred on day 1 of even-numbered cycles. Imaging was obtained every 8 weeks and response assessed using the Response Evaluation Criteria in Solid Tumours (RECIST) guidelines version 1.0 (Therasse et al, 2000).
Pharmacokinetic sampling, assay, and analysis
During cycle 1, blood samples for the analysis of paclitaxel were obtained before treatment and at 0.5, 1 (end of infusion), 1.5, 2, 2.5, 3, 4, 5, 6, 8, 10, and 24 h after the start of the infusion on days 1 and 15 (pazopanib and lapatinib were withheld on day 1). Also during cycle 1, blood samples were collected to determine plasma pazopanib and lapatinib concentrations before dosing and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, and 24 h post dosing on day 15. High-performance liquid chromatography and tandem mass spectrometry were used to quantify the plasma concentrations of paclitaxel, pazopanib, and lapatinib. Paclitaxel was extracted from 0.1 ml of plasma by a liquid–liquid technique, with a lower limit of quantitation of 2 ng ml−1. Pazopanib and lapatinib were extracted from plasma by a liquid–liquid method; validated concentration ranges were 100–50 000 ng ml−1 for pazopanib and 5–5000 ng ml−1 for lapatinib.
Analysis of pharmacokinetic parameters included Cmax, AUC from time of dosing to last time of measurable concentration (AUC0–t) or extrapolated to infinity (AUC0–∞), systemic clearance, apparent volume of distribution (Vss), and terminal half-life (t1/2) of paclitaxel when administered alone and in combination with pazopanib and lapatinib, and AUC0–t, AUC from time of dose to 24 h (AUC0–24), and Cmax of pazopanib and lapatinib when administered with other study drugs. Pharmacokinetic parameters were analysed by standard noncompartmental analysis using WinNonlin Pro version 4.1 or higher (Pharsight Corporation, Mountain View, CA, USA). The effect of pazopanib and lapatinib on the pharmacokinetics of paclitaxel was tested statistically by performing an analysis of variance on data from patients at the MTR.
Results
Patient Characteristics
A total of 26 patients were enrolled between July 2007 and April 2009 (Table 1). The most common tumour types were biliary tract cancer (23%), non-small cell lung cancer (12%), and salivary gland cancer (12%). Approximately 50% of the patients had received less than two lines of previous chemotherapy. No patients are currently on study. Nine patients had received prior taxane therapy (either paclitaxel or docetaxel).
Dose escalation and dose-limiting toxicity
The starting dose level for paclitaxel, pazopanib, and lapatinib was 50 mg m−2, 400 mg per day, and 1000 mg per day, respectively (Table 2). Paclitaxel starting doses were selected so that the expected systemic exposure of paclitaxel at the selected dose level would not exceed 115% of the predicted exposure at 100 mg m−2. One of the initial three patients experienced a DLT of grade 3 elevation in ALT and AST, which led to a 2-week delay in starting cycle 2. Dose level 1 was expanded, and none of the additional evaluable patients had a DLT. One patient was not evaluable for DLT because a prohibited medication had been taken; this patient was withdrawn from the study and replaced. Because one of six evaluable patients at dose level 1 experienced a DLT, enrollment escalated to dose level 2, in which the pazopanib dose was increased and the paclitaxel dose was maintained. Three patients were treated with paclitaxel 50 mg m−2, pazopanib 800 mg per day, and lapatinib 1000 mg per day. There were no DLTs during the first cycle. At dose level 3, the paclitaxel dose was increased to 80 mg m−1, and pazopanib and lapatinib were maintained at 800 mg per day and 1000 mg per day, respectively. Two of the three patients experienced DLTs of grade 4 neutropenia and grade 3 fatigue. It was deemed useful to evaluate the three-drug combination with a lower dose of pazopanib at 400 mg in dose level 4. The first three evaluable patients at dose level 4 were treated with paclitaxel 80 mg m−2, pazopanib 400 mg per day, and lapatinib 1000 mg per day, and completed cycle 1 with no DLTs. Given the toxicities observed at dose level 3, it was decided to enroll additional patients at dose level 4 to further assess the tolerability. Among the nine evaluable patients, there were two DLTs, including one patient with grade 4 thrombosis and another patient with elevations of ALT (grade 2) and AST (grade 3) that delayed the start of cycle 2 by more than 2 weeks. There was one non-evaluable patient for whom a replacement was needed at dose level 4. Overall, two of the twelve evaluable patients experienced a DLT, and given these findings, the MTR was determined to be paclitaxel 80 mg m−2 on days 1, 8, and 15 every 28 days with pazopanib 400 mg per day and lapatinib 1000 mg per day.
Toxicities
The most common treatment-related adverse events (all grades) were diarrhoea (54%), fatigue (50%), neutropenia (50%), nausea (38%), and anorexia (35%) across all 4 dose levels (Table 3). The majority of toxicities were grade 1 or 2. The most common treatment-related toxicities at the MTR (dose level 4) were diarrhoea (69%), fatigue (46%), nausea (31%), and anorexia (31%). Twenty patients (77%) experienced adverse events leading to dose interruption of study drugs after cycle 1. The most common reasons were neutropenia (23%), increased ALT and AST (12% each), nausea (12%), and diarrhoea (12%). Two patients at the MTR experienced diarrhoea that led to an interruption in study drug dosing. Elevations in liver function tests, including AST, ALT, bilirubin, and alkaline phosphatase, were reported (Table 3). There were no grade 4 hepatobiliary events. Five patients experienced at least one hepatobiliary event that led to an interruption of dosing. Two patients had to discontinue study drugs and withdraw from study due to grade 2 AST/ALT elevation and grade 2 hyperbilirubinemia that were thought to be related to treatment. The most common cardiovascular toxicity was hypertension. Sixteen patients had baseline hypertension. Six of these patients required additional antihypertensive agents and five patients required dose adjustments of their antihypertensive medications. One patient had grade 3 hypertension at the MTR. There were no episodes of significant QTc prolongation, which was defined as >500 msec. There was an occurrence of grade 3 large-intestine perforation that was considered treatment-related. There were no major haemorrhagic events that were thought to be related to the study regimen. One grade 4 thrombosis occurred at dose level 4. One grade 2 hypothyroidism occurred at dose level 2. No proteinuria was reported during the study. Grade 4 haematologic toxicity consisted mostly of neutropenia and thrombocytopenia. At dose level 3, one patient experienced both grade 4 neutropenia and grade 4 thrombocytopenia. At dose level 4, grade 4 neutropenia occurred in two patients. Four deaths occurred during the study, after treatment was discontinued. These deaths were not considered study-related, but due to the underlying disease.
Pharmacokinetics
Plasma samples for analysis of the pharmacokinetics of paclitaxel in the presence and absence of pazopanib and lapatinib at the MTR were obtained on day 1 (n=11) and on day 15 (n=9). Systemic exposure to pazopanib did not increase in a dose-proportional fashion. A two-fold increase in pazopanib dose (400–800 mg per day) resulted in a less than two-fold increase in mean AUC0–24 and mean Cmax (Table 4). Increasing the paclitaxel dose from 50–80 mg m−2 had little effect on the observed AUC0–24 and Cmax of pazopanib. The daily lapatinib dose (1000 mg) did not change during the study; plasma lapatinib concentrations were measured to determine if changes in dosing of other study drugs altered its pharmacokinetic parameters. For patients receiving paclitaxel 50 mg m−2, doubling the pazopanib dose led to a 2.6-fold increase in AUC0–24 and a 2.9-fold increase in the Cmax of lapatinib. For patients receiving pazopanib 400 mg, increasing paclitaxel from 50 to 80 mg m−2 led to increases in the AUC0–24 and Cmax of lapatinib (1.4- and 1.5-fold, respectively). However, for the one evaluable patient at the highest dosing level (paclitaxel 80 mg m−2, pazopanib 800 mg, and lapatinib 1000 mg), AUC0–24 and Cmax values of lapatinib were lower than the mean values recorded for all other dose levels.
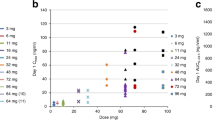
Pharmacokinetic parameters for paclitaxel on days 1 and 15 are reported in Table 5. At all dose levels where mean or median values were available, paclitaxel clearance was lower on day 15 of cycle 1 than on day 1; AUC0–∞ and Cmax were higher on day 15. At the MTR of pazopanib 400 mg, lapatinib 1000 mg, and paclitaxel 80 mg m−2, median Cmax (Figure 1) and AUC0–24 of paclitaxel were higher on day 15 than on day 1. Statistical analysis revealed that coadministration of pazopanib and lapatinib with paclitaxel at the MTR resulted in a 38% higher geometric least-squares mean Cmax of paclitaxel than with administration of paclitaxel alone (Cmax ratio (90% confidence interval): 1.38 (1.19, 1.59)).

Median paclitaxel concentration in the absence (day 1) or presence (day 15) of lapatinib and pazopanib at the MTR (paclitaxel 80 mg m −2 , pazopanib 400 mg, and lapatinib 1000 mg).
Efficacy
One partial response was reported in a patient with salivary gland tumour who had received prior therapy with paclitaxel and carboplatin and who received 10 cycles of study treatment at dose level 2. This patient achieved a partial response after 3 cycles of treatment. Nine patients had stable disease, with three patients experiencing disease stabilisation for 6 months or more; the tumour types included oesophageal cancer (treated at dose level 1), cholangiocarcinoma (treated at dose level 4), and adenocarcinoma of unknown primary (treated at dose level 1). Among these three patients, only the patient with adenocarcinoma of unknown primary had prior taxane therapy with paclitaxel. There were two breast cancer patients, one with a triple negative tumour and the other with HER2-positive disease; neither patient experienced a response.
Discussion
In this phase I study, we sought to determine the MTR of the combination of weekly paclitaxel with two biologically targeted agents, pazopanib and lapatinib. Pazopanib 400 mg and lapatinib 1000 mg were chosen as reasonable starting doses for dose level 1, with the understanding that pazopanib and lapatinib, both CYP3A4 inhibitors, would result in increased exposure to paclitaxel, a CYP3A4 substrate. In addition, the previously reported phase I study of pazopanib and lapatinib determined the recommended phase II dose of pazopanib to be 800 mg and of lapatinib to be 1500 mg. This study did not detect any drug–drug interactions between either compound at pazopanib 400 mg and lapatinib 1000 mg, supporting the starting doses for dose level 1 (de Jonge et al, 2013).
We determined the MTR to be paclitaxel 80 mg m−2 on days 1, 8, and 15 every 28 days with pazopanib 400 mg per day and lapatinib 1000 mg per day. This includes a lower pazopanib dose than the approved 800 mg per day monotherapy dose and a lower lapatinib dose than that used in the monotherapy phase II studies, but a clinically acceptable dose of weekly paclitaxel. Our previous phase I study of pazopanib in combination with paclitaxel reported a 36% increase in the Cmax of paclitaxel and a 26% increase in the geometric mean AUC relative to administration of paclitaxel alone (Tan et al, 2010). Our current study results show that with the addition of lapatinib, concomitant administration of paclitaxel and pazopanib at the MTR resulted in an approximately 38% increase in the geometric least-squares mean paclitaxel Cmax and an approximately 37% increase in the geometric mean paclitaxel AUC, relative to administration of paclitaxel 80 mg m−2 alone. The increase in paclitaxel AUC and Cmax was less than two-fold in the presence of pazopanib and lapatinib, indicating that both are weak inhibitors of CYP3A4 and CYP2C8. As expected, an increase in the paclitaxel dose had no apparent effect on the pharmacokinetics of pazopanib. An increase in the pazopanib dose from 400 mg per day to 800 mg per day led to an increase in Cmax and exposure of lapatinib in patients receiving paclitaxel 50 mg m−2. Because paclitaxel 80 mg m−2 with pazopanib 800 mg achieved higher systemic exposure of paclitaxel compared with paclitaxel administered alone, consideration could have been given to evaluating an intermediate dose of paclitaxel between 50 mg m−2 and 80 mg m−2, such as paclitaxel 60 mg m−2, with pazopanib 800 mg per day and lapatinib 1000 mg per day, but an intermediate dose was not evaluated. Real-time pharmacokinetic analysis of paclitaxel after patients completed the first cycle of treatment may have been able to guide the protocol-dosing levels.
The most common adverse events with the three-drug combination were diarrhoea, neutropenia, fatigue, nausea, and anorexia. This is consistent with the toxicity profile of each agent alone. The majority of the most common nonhematologic toxicities was grade ⩽2, whereas the majority of neutropenia events was grade ⩾3. Overall, the frequency of neutropenia (all grades) in this study was 50%, higher than that reported for weekly paclitaxel plus pazopanib (23%) (Tan et al, 2010). This may be attributed to the increase in systemic exposure of paclitaxel relative to the administration of paclitaxel alone after concomitant administration of pazopanib and lapatinib as shown by our pharmacokinetic results. Also, the rate of grade 3 drug-related neutropenia at the MTR (4 of 13 patients, 31%) was higher than that reported in a previous phase I trial of lapatinib 1500 mg per day and paclitaxel 80 mg m−2 weekly (1 of 12 patients, 8%); however, the rate of grade 3 drug-related diarrhoea at the MTR in this trial (2 of 13 patients, 15%) was three-fold lower than that reported in the previous trial (6 of 12 patients, 50%) (Jones et al, 2004; Crown et al, 2007). This finding suggests that the lower lapatinib dose of 1000 mg per day in the MTR may be associated with a lower frequency of diarrhoea.
This triplet combination is most clinically relevant in the treatment of patients with HER2-positive metastatic breast cancer. Preclinical data demonstrate that increased expression of VEGF is associated with HER2 overexpression and is a downstream target of the HER2-signalling pathway, which supports combining the inhibition of HER2 and VEGF in the treatment of HER2-positive breast cancers (Konecny et al, 2004). Only two breast cancer patients were enrolled in our trial and only one patient was HER2-positive; thus, the number of breast cancer patients is too small to draw any conclusion about the antitumor activity of this regimen.
In summary, the concurrent administration of pazopanib and lapatinib with paclitaxel caused an increase in the paclitaxel AUC and Cmax, which may explain in part the higher rate of neutropenia than has been demonstrated with paclitaxel–pazopanib and paclitaxel–lapatinib combinations. However, weekly paclitaxel on days 1, 8, and 15 every 28 days in combination with pazopanib 400 mg and lapatinib 1000 mg once daily can be given safely. Targeting the VEGF, EGF, and HER2 receptors simultaneously with chemotherapy needs further exploration to determine the specific tumour types for which patients might derive improved therapeutic benefit.
Change history
27 May 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Abad M, Calvo I, Martinez N, Herreo M, Quijano Y, Duran H, Aranda M, Suarez A, Lopez-Rios F, Perez D, Perea S, Hidalgo M, Garcia-Estevez L (2012) Neoadjuvant bevacizumab and trastuzumab in combination with weekly paclitaxel as neoadjuvant treatment in HER2-positive breast cancer: results from a phase II trial (AVANTHER). J Clin Oncol 30 (15S): Abstract 602.
Cristofanilli M, Johnston SR, Manikhas A, Gomez HL, Gladkov O, Shao Z, Safina S, Blackwell KL, Alvarez RH, Rubin SD, Ranganathan S, Redhu S, Trudeau ME (2013) A randomized phase II study of lapatinib+pazopanib versus lapatinib in patients with HER2+ inflammatory breast cancer. Breast Cancer Res Treat 137 (2): 471–482.
Crown JP, Burris HA III, Jones S, Koch KM, Fittipaldo A, Parikh R, Koehler M (2007) Safety and tolerability of lapatinib in combination with taxanes (T) in patients with breast cancer (BC). J Clin Oncol 25 (18S): Abstract 1027.
de Jonge MJ, Hamberg P, Verweij J, Savage S, Suttle AB, Hodge J, Arumugham T, Pandite LN, Hurwitz HI (2013) Phase I and pharmacokinetic study of pazopanib and lapatinib combination therapy in patients with advanced solid tumors. Invest New Drugs 31 (3): 751–759.
Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello-Gruszfeld A, Crown J, Chan A, Kaufman B, Skarlos D, Campone M, Davidson N, Berger M, Oliva C, Rubin SD, Stein S, Cameron D (2006) Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 355 (26): 2733–2743.
Harris PA, Boloor A, Cheung M, Kumar R, Crosby RM, Davis-Ward RG, Epperly AH, Hinkle KW, Hunter RN, Johnson JH, Knick VB, Laudeman CP, Luttrell DK, Mook RA, Nolte RT, Rudolph SK, Szewczyk JR, Truesdale AT, Veal JM, Wang L, Stafford JA (2008) Discovery of 5-((4-((2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl)amino)-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem 51 (15): 4632–4640.
Johnston S, Pippen J, Pivot X, Lichinitser M, Sadeghi S, Dieras V, Gomez HL, Romieu G, Manikhas A, Kennedy MJ, Press MF, Maltzman J, Florance A, O'Rourke L, Oliva C, Stein S, Pegram M (2009) Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol 27 (33): 5538–5546.
Johnston SR, Gómez H, Stemmer SM, Richie M, Durante M, Pandite L, Goodman V, Slamon D (2013) A randomized and open-label trial evaluating the addition of pazopanib to lapatinib as first-line therapy in patients with HER2-positive advanced breast cancer. Breast Cancer Res Treat 137 (3): 755–766.
Jones SF, Burris HA, Yardley DA, Greco FA, Spigel DR, Raefsky EL, Hainsworth JD, Willcutt NT, Calvert SA, Versola MJ (2004) Lapatinib (an oral dual kinase inhibitor) plus weekly or every 3 week paclitaxel. Breast Cancer Res Treat 88 (suppl 1): Abstract 1069.
Konecny GE, Meng YG, Untch M, Wang HJ, Bauerfeind I, Epstein M, Stieber P, Vernes JM, Gutierrez J, Hong K, Beryt M, Hepp H, Slamon DJ, Pegram MD (2004) Association between HER-2/neu and vascular endothelial growth factor expression predicts clinical outcome in primary breast cancer patients. Clin Cancer Res 10 (5): 1706–1716.
Konecny GE, Pegram MD, Venkatesan N, Finn R, Yang G, Rahmeh M, Untch M, Rusnak DW, Spehar G, Mullin RJ, Keith BR, Gilmer TM, Berger M, Podratz KC, Slamon DJ (2006) Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res 66 (3): 1630–1639.
Lau D, Guo L, Gandara D, Young LJ, Xue L (2004) Is inhibition of cancer angiogenesis and growth by paclitaxel schedule dependent? Anticancer Drugs 15 (9): 871–875.
Olaussen KA, Commo F, Tailler M, Lacroix L, Vitale I, Raza SQ, Richon C, Dessen P, Lazar V, Soria JC, Kroemer G (2009) Synergistic proapoptotic effects of the two tyrosine kinase inhibitors pazopanib and lapatinib on multiple carcinoma cell lines. Oncogene 28 (48): 4249–4260.
Rubin BP, Duensing A (2006) Mechanisms of resistance to small molecule kinase inhibition in the treatment of solid tumors. Lab Invest 86 (10): 981–986.
Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson DH (2006) Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 355 (24): 2542–2550.
Shaked Y, Henke E, Roodhart JM, Mancuso P, Langenberg MH, Colleoni M, Daenen LG, Man S, Xu P, Emmenegger U, Tang T, Zhu Z, Witte L, Strieter RM, Bertolini F, Voest EE, Benezra R, Kerbel RS (2008) Rapid chemotherapy-induced acute endothelial progenitor cell mobilization: implications for antiangiogenic drugs as chemosensitizing agents. Cancer Cell 14 (3): 263–273.
Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, Barrios CH, Salman P, Gladkov OA, Kavina A, Zarbá JJ, Chen M, McCann L, Pandite L, Roychowdhury DF, Hawkins RE (2010) Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 28 (6): 1061–1068.
Tan AR, Dowlati A, Jones SF, Infante JR, Nishioka J, Fang L, Hodge JP, Gainer SD, Arumugham T, Suttle AB, Dar MM, Lager JJ, Burris HA (2010) Phase I study of pazopanib in combination with weekly paclitaxel in patients with advanced solid tumors. Oncologist 15 (12): 1253–1261.
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92 (3): 205–216.
van der Graaf WT, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, Casali PG, Schoffski P, Aglietta M, Staddon AP, Beppu Y, Le Cesne A, Gelderblom H, Judson IR, Araki N, Ouali M, Marreaud S, Hodge R, Dewji MR, Coens C, Demetri GD, Fletcher CD, Dei Tos AP, Hohenberger P EORTC Soft Tissue and Bone Sarcoma Group; PALETTE study group (2012) Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 379 (9829): 1879–1886.
Acknowledgements
We thank Dr Mohammed M. Dar for his contributions to this study. Medical editorial assistance was provided by William Sinkins, PhD, at ProEd Communications, Beachwood, Ohio. This study (NCT00388076) was sponsored and funded by GlaxoSmithKline Pharmaceuticals, Philadelphia, Pennsylvania. Editorial assistance was also supported by GlaxoSmithKline.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
ART has received research funding from GlaxoSmithKline. JRI reports a consultant/advisory role (uncompensated). ABS is a GlaxoSmithKline employee and stockholder. AD, MNS, SFJ, JB, MPK, KTL, and HABIII report no potential conflict of interest associated with this publication.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Tan, A., Dowlati, A., Stein, M. et al. Phase I study of weekly paclitaxel in combination with pazopanib and lapatinib in advanced solid malignancies. Br J Cancer 110, 2647–2654 (2014). https://doi.org/10.1038/bjc.2014.233
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.233
Keywords
This article is cited by
-
Pazopanib Monotherapy Is Active in Relapsed and Refractory Metastatic Gastroesophageal Adenocarcinoma and Can Produce Durable Response
Journal of Gastrointestinal Cancer (2019)
-
Pharmacodynamic and Pharmacokinetic Markers For Anti-angiogenic Cancer Therapy: Implications for Dosing and Selection of Patients
European Journal of Drug Metabolism and Pharmacokinetics (2018)
-
A Phase I, Dose-Escalation Trial of Pazopanib in Combination with Cisplatin in Patients with Advanced Solid Tumors: A UNICANCER Study
Oncology and Therapy (2016)
