
Abstract
Background:
Androgen receptor (AR)-gene amplification, found in 20–30% of castration-resistant prostate cancer (CRPCa) is proposed to develop as a consequence of hormone-deprivation therapy and be a prime cause of treatment failure. Here we investigate AR-gene amplification in cancers before hormone deprivation therapy.
Methods:
A tissue microarray (TMA) series of 596 hormone-naive prostate cancers (HNPCas) was screened for chromosome X and AR-gene locus-specific copy number alterations using four-colour fluorescence in situ hybridisation.
Results:
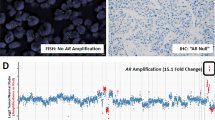
Both high level gain in chromosome X (⩾4 fold; n=4, 0.7%) and locus-specific amplification of the AR-gene (n=6, 1%) were detected at low frequencies in HNPCa TMAs. Fluorescence in situ hybridisation mapping whole sections taken from the original HNPCa specimen blocks demonstrated that AR-gene amplifications exist in small foci of cells (⩽600 nm, ⩽1% of tumour volume). Patients with AR gene-locus-specific copy number gains had poorer prostate cancer-specific survival.
Conclusion:
Small clonal foci of cancer containing high level gain of the androgen receptor (AR)-gene develop before hormone deprivation therapy. Their small size makes detection by TMA inefficient and suggests a higher prevalence than that reported herein. It is hypothesised that a large proportion of AR-amplified CRPCa could pre-date hormone deprivation therapy and that these patients would potentially benefit from early total androgen ablation.
Similar content being viewed by others

Main
Prostate cancer (PCa) has a highly variable natural history. The available management options for patients diagnosed with early-stage disease vary from deferred treatment (active surveillance) to treatments with significant morbidity (radiotherapy/surgery). Biomarkers are urgently required to risk-stratify patients so that those with low-risk disease might avoid unnecessary treatment. Conversely, patients initially diagnosed with high-risk disease could be targeted for more aggressive first-line therapy. To identify such biomarkers, the TransAtlantic Prostate Group established a cohort of PCa patients whose disease was conservatively managed and where samples for biomarker analysis were taken before hormone withdrawal therapy. In fluorescence in situ hybridisation (FISH)-based analyses, this clinical cohort has already yielded the discovery that two copies of a TMPRSS2/ERG translocation are associated with extremely poor PCa-specific survival (Attard et al, 2008; Clark et al, 2008), and that PTEN gene loss in the absence of ERG or ETV1 gene rearrangements identifies a group of poor prognosis patients (Reid et al, 2010).
Proliferation and survival of PCa cells are critically dependent on androgen stimulation, with treatment being primarily based on conventional androgen deprivation (also referred to as castration). Such treatments can often result in dramatic remission. However, the disease invariably relapses forming castration-resistant PCa (CRPCa), which features hypersensitivity of androgen pathway signalling that may result from amplification or activating mutations of the AR gene. AR-gene mutation has been reported in around 20% (Taplin et al, 1995, 2003) and AR amplification in 20–30% of CRPCa (Koivisto et al, 1997; Linja et al, 2001; Haapala et al, 2007), and has been suggested to develop during hormone deprivation therapy (Visakorpi et al, 1995; Palmberg et al, 1997; Haapala et al, 2007; Waltering et al, 2012). In contrast, AR-gene amplification has been reported at a low frequency in primary hormone naive cancers (1%, 3 out of 293 tumours from five studies; Koivisto et al, 1997; Palmberg et al, 1997; Bubendorf et al, 1999; Brown et al, 2002; Edwards et al, 2003). However, these published studies were limited by their use of small patient numbers, minimal sampling strategies (e.g., a single 0.6 mm core extracted from an entire cancer (Bubendorf et al, 1999), no patient survival data, high Gleason samples (Koivisto et al, 1997; Edwards et al, 2003) and a basic definition of the AR-gene alteration using single colour AR FISH probes. A recent study by Taylor et al (2010) using gene copy number data from Agilent CGH arrays found no AR amplification in 181 primary tumours. Knowledge of the timing of occurrence of AR-gene amplification is critical. If such alterations are indeed induced by androgen withdrawal, they may have limited relevance to first-line patient therapy. However, if they are present before therapy, they could help inform on patient treatment.
Here we have used four-colour FISH probes to characterise the diversity of AR-gene alterations in human prostate cancer (PCa), and to assess their occurrence and clinical significance in a series of 596 hormone naive cancers represented on a TMA by up to six cores per patient and linked to 10 years follow-up survival data. We have in addition interrogated a number of whole-block tissue sections to investigate the prevalence of AR copy number altered PCa within individual selected samples.
Materials and Methods
Patients
Samples were collected by the TransAtlantic Prostate Group as previously described (Cuzick et al, 2006), briefly, patients were included if they were under 76 years at diagnosis and had clinically localised PCa diagnosed by needle biopsy or transurethral resection of the prostate. A number of exclusion criteria were used to focus the study on patients who were likely to have truly localised disease at presentation: patients were excluded within 6 months of diagnosis if they had been treated by radical prostatectomy or radiation therapy or had objective or clinical evidence of metastatic disease or a PSA measurement >100 ng ml−l. In addition, those with objective evidence of metastatic disease (by bone scan, X-ray, radiograph, CT scan, MRI, bone biopsy, lymph node biopsy, pelvic lymph node dissection) or clinical indications of metastatic disease (including pathologic fracture, soft tissue metastases, spinal compression or bone pain) were also excluded. Men who had had hormone therapy before diagnostic biopsy were also excluded, as were men who died within 6 months of diagnosis or had <6 months follow-up. The Northern Multi-Research Ethics Committee provided national approval followed by local ethics committee approval at each of the collaborating hospitals.
Tissue microarrays (TMAs)
Tissue microarrays were constructed as described by Attard et al (2008) with up to six cores of 600 μm diameter taken from each tumour. Reassignment as either ‘cancer’ or ‘normal’ was by histopathological examination of H&E-stained sections flanking the TMA slice used for FISH. Fluorescence in situ hybridisation was scorable in 596 of 808 patient samples.
Fluorescence in situ hybridisation studies
Fluorescence in situ hybridisation probe and TMA preparation were carried out as previously reported (Clark et al, 2008). See Figure 1A and legend for FISH probes at the AR-locus. The AR FISH scores were divided into five categories (see Figure 1B for examples and a description of the ‘GBRX’ (Green/Blue/Red/cenX) counting system). All areas of each TMA core were scored, where multiple AR patterns of gain and amplification were present, the highest AR copy number visible in a minimum of 50 cells was recorded as the score in each core or patient. The term ‘amplification’ was used to describe any increase in AR-gene copy number.

(A) Multi-coloured fluorescence in situ hybridisation (FISH) probes used for investigating androgen receptor ( AR ) gene locus copy number (relative probe positions shown and actual distances indicated): (i) blue probe spanning the AR-gene (blue arrow) consisted of bacterial artificial chromosomes (BACs) RP11-479J1, RP11-963N10, CTD-2155B10 labelled with Aqua PlatinumBright kit (Kreatech Diagnostics). (ii) Red probe telomeric to AR, BACs RP11-466E18 and RP11-768G22 visualised with Cy3, (iii) Green probe centromeric to AR, BACs RP11-414C19 and RP11-745E2 visualised with FITC. Chromosome X (ChrX) centromere probe (CenX) was a 500-bp PCR product as Warburton et al (1991), sonicated and directly labelled with PlatinumBright 647 kit (far-red; Kreatech Diagnostics). (B) Examples of the five categories (Cat1–5) of AR copy number alterations. Cat1–4 consisted of increasing numbers of ChrX: Cat5 consisted of locus-specific amplification of AR. We scored the tumours by counting the Green, Blue, Red and CenX signals. For example, Cat1 contain a single ChrX in normal male cells seen as single overlapping Green (G), Blue (B) and Red (R) spots, which numerically correlated with a single centromere X signal (X) (not shown). This normal male pattern was counted as 1 : 1 : 1 : 1 for the numbers of GBRX probes counted in a cell. Gain of the ChrX was inferred when multiples of this pattern were seen (Cat2–4). In contrast, Cat5 amplification of the AR-gene locus could be visualised as strings of coloured beads corresponding to the co-amplification of different combinations of the Green, Blue and Red signals. Amplifications could involve all three AR locus colours or be just multiple adjacent copies of the Green and Blue signals or Blue and Red signals (as shown here). Amplification of the Blue probe alone was not seen in these studies.
Statistical analysis
The primary end points for this study were time to death from PCa and time to death from any cause. Univariate and multivariate analysis were carried out using proportional hazard (Cox) regression analysis (Cox and Oakes, 1984). All follow-up times commenced at the point of 6 months following diagnosis. Associations between categorical data were examined using the χ2-test, χ2-test for trend and Fisher’s exact test when expected cell counts were less than 5. Associations between categorical and numerical variables were assessed using analysis of variance. All P-values were two-sided. The following variables, determined as described previously (Cuzick et al, 2006; Berney et al, 2009), were included in the multivariate analyses: centrally reviewed Gleason score, Ki-67, baseline PSA (last PSA value within 6 months of diagnosis) and age at diagnosis.
Results
AR-gene status in hormone naive prostate cancer (HNPCa)
To assess AR-gene copy number gain and amplification, we used a multi-colour probe system consisting of three differentially labelled probes at the AR-locus (see Figure 1) plus an X-Centromere probe to determine ChrX copy number. The observed AR status of each patient was assigned to one of five categories (Cat) depending on the ChrX copy number (categories 1–4) or the presence of AR-gene locus-specific copy number gain (Cat5; see Figure 1).
AR FISH status was determined for a TMA series of 596 patients that had not been treated with androgen withdrawal therapy before sample collection (Cuzick et al, 2006) Cat1 (n=444), Cat2 (n=127), Cat3 (n=15) and Cat4 (n=four). AR-locus-specific (Cat5) amplification was observed in six patients; three of which had gain of more than five copies of AR, one had three copies and two exhibited duplications. Correlations with clinical variables demonstrated an association between AR category and increased Gleason score, increased baseline PSA and the percentage of cancer in the original diagnostic biopsy (P<0.001 for each of these factors), and also with more advanced clinical stage (P=0.025; Table 1). There was no significant difference in age between the different categories (P=0.550).
Locus-specific amplification and high copy number AR gain correlates with poorer cancer-specific survival
Correlations with outcome were first considered for patients in categories 1 to 4. Univariate analysis (Table 2) demonstrated consistently poorer PCa-specific survival in patients with an increased ChrX copy number when compared with patients with a single copy of ChrX. However, this association was not maintained in multivariate analyses that included Gleason score, PSA level, patient age and cancer Ki67 status (Table 2). General aneuploidy has been linked to poorer PCa survival (Taylor et al, 2010), it is not known whether the chromosome X gains were specific to X or a general genomic gain.
Locus-specific amplification (Cat5) was significantly associated with poorer PCa-specific survival (hazard ratio (HR)=5.08 (95% CI=1.85–13.95)) in univariate analysis when compared with patients with a single copy of the AR-gene (Figure 2). The association became more marked in multivariate analyses that took into account Gleason sum, PSA, age and Ki67 (HR=10.73 (95% CI=3.64–31.66)). In analyses of overall mortality, Cat5 alterations exhibited a non-significant trend towards worse outcome (HR=1.61, 95% CI=0.60–4.32) that became significant in multivariate analysis (HR=3.71, 95% CI=1.35–10.18). Overall, the results demonstrated a consistent correlation between AR copy number and poorer cancer-specific survival; however, because of the small number of Cat5 patients in these analyses (n=6), the results involving analyses of these particular samples were considered to be anecdotal only.

Kaplan–Meier plot of prostate cancer-specific mortality for the five AR FISH categories (Cat1–5, see Figure 1 for AR scoring explanation).
Focal origin of locus-specific AR-gene amplification
For each of the Cat5 samples, only a single core of the 1–4 cores assessed by FISH exhibited locus-specific AR-gene amplification (Table 3) demonstrating heterogeneity of AR status in cancer from single prostates. In addition, one of the cores assigned to Cat5 (Table 3; sample 4, core 2) contained two adjacent areas of high copy number gain: one with Cat5 (GBRX/2552) and one with Cat4 containing four to six copies of the X chromosome (Figure 3). This indicated that distinct mechanisms of high-copy number AR gain can occur in the same cancer.

Multiple patterns of AR gain and amplification found in a single TMA core. (A) Cat5 AR locus-specific amplification to the left of the yellow dotted line; (B) Cat4 ChrX gain to the right (CenX probe not shown; sample 4, TMA core 2 in Table 3).
To investigate these observations further, sections were cut from Cat5 whole-block TURP samples where significant amounts of tissue remained (samples 3, 4 and 5, Table 3). AR FISH maps for samples 3 and 4 are shown in Figure 4A and B. For sample 4, we detected small regions (foci) of cells with Cat5 and Cat4 alterations: both of these foci had been selected in the TMA cores (Table 3). The Cat5 alterations represented about 1% in the cancer areas in this TURP specimen and had the same GBRX FISH pattern as found in the TMA core. For samples 3 and 5, we failed to detect areas of Cat5 cancer, although two small areas of Cat4 cancer were observed in sample 3.

AR FISH analysis of whole TURP-block sections of samples 3, 4 and 7 (see Table 3 ). Coloured areas indicate the FISH score in areas of tumour identified by pathologist examination of an adjacent H&E-stained slice. Non-coloured areas are not tumour and were not scored. White areas are Cat1 with a single ChrX. Yellow and green circles indicate positions of the 600 μm diameter cores taken from areas of tumour and normal, respectively, for TMA construction. Blue and red arrows highlight the small foci of Cat4 and Cat5 tumour, respectively. The nuclei pictures in the key area are from sample 7. (A) Sample 4, cores taken for TMA construction have by chance sampled both the Cat4 and Cat5 tumour areas; (B) sample 3, this sample had a Cat5 tumour on TMA analysis, however, no Cat5 tumour was visible on whole-block analysis but foci of Cat4 tumour were present; (C) sample 7, this sample had only a single AR-amplified cell on TMA analysis but on whole-block analysis both Cat4 and Cat5 tumour foci were found. In all cases, Cat5 tumour foci were less than 1% of the whole-block tumour area.
In the TMA analysis, we also detected three additional cases that exhibited between one and four cells containing a locus-specific AR-gene amplification (see example in Supplementary Figure 1). These cases were not assigned Cat5 status because they did not meet the scoring threshold of containing at least 50 altered cells. For example, sample 7 in Table 3 had three to five copies of ChrX on TMA analysis and had been scored as Cat4. However, in core four, a single cell was observed that had a locus-specific AR-gene amplification. The AR FISH map for this TURP specimen is shown in Figure 4C. Over 99% of the cancer contained either two copies or three copies of ChrX. In addition four small areas contained higher copy number gains. One focus harboured five copies of ChrX, whereas three other foci contained Cat5 AR-locus-specific amplifications of >seven-fold. None of these areas of Cat5 amplification were near the sites selected for TMA cores. Survival time for this patient was 4.2 years. Cat5 foci were not detected in the other two whole-block TURP sections, which were still alive at last follow-up (12 and 14 years).
The presence of Cat4 and Cat5 alterations appear to be linked. Only four Cat4 samples were found in TMA analyses (0.7%), however, when combining TMA and whole TURP analysis, three of the seven samples containing Cat5 alterations (3, 4 and 7) also had areas of Cat4 cancer (see Table 2 and Figure 4). In two of these samples, foci of Cat4 and Cat5 foci were in immediately adjacent tumour areas (Figures 3 and 4C).
From these observations, we draw several conclusions. First, foci of cells harbouring locus-specific AR-gene amplification occur in fields of tumour containing ChrX copy number gain. Second, high copy number gains of ChrX (Cat4) and locus-specific gains in AR frequently occur together in separate regions of the same cancer. Third, in the cases that we examined, foci of cells containing Cat4 and Cat5 alterations represent only a small percentage (<1%) of the overall cancer cell population.
Discussion
The genetically heterogeneous nature of cancer is well documented. However, different models exist to explain its origin and significance. One view is that cancer clones can be arranged in a hierarchical structure that is maintained by rare (∼1 in 106) ‘tumour-propagating cells’, also called ‘cancer stem cells’. An alternative view is that a significant proportion (or even the majority) of individual cells that have undergone malignant transformation can propagate a cancer (Quintana et al, 2008; Shackleton et al, 2009). In the latter model, even small volume clones arising within a tumour may be significant if they possess a growth advantage. In breast cancer, inter-tumoural heterogeneity of HER2 amplification linked to HER2 overexpression has been documented, and evidence supports the idea that tumour progression may in some cases result from selection of HER2 overexpressing subclones present in the primary cancer (Cottu et al, 2008). We now hypothesise that a similar model of progression arises in PCa: namely that subclones of cells harbouring AR gain/amplification and consequent overexpression arise in primary hormone naive tumours. AR amplification could, in principle, be due to conditions of localised androgen insufficiency within a PCa as a result of, for example, poor tumour vascularisation, or a general requirement for increased androgen signalling in tumour cells, which could be growth limiting to the tumour. We propose that these clonal growths would have a survival advantage on initiation of hormone deprivation therapy, and could seed the formation of CRPC, with concurrent further amplification of the AR gene by similar mechanisms (Figure 5, route 1). This has implications for both continuous and intermittent androgen deprivation therapy (Salonen et al, 2013). In support of this concept, the current study has demonstrated the occurrence of AR-gene amplification in HNPCa and revealed a consistent association between AR copy number and poor cancer-specific survival. Previously, it has been shown that increases in AR transcript level (a change that has been associated with AR amplification (Koivisto et al, 1997; Linja et al, 2001; Edwards et al, 2003)) are the only consistent alteration associated with the development of resistance to castration, and it has been demonstrated that increasing levels of AR can confer resistance to castration by amplifying signal output from low levels of residual ligand (Waltering et al, 2009). In principle, clones of cells containing AR amplification could exhibit a growth advantage and gradually become the predominant cancer clone, even in the absence of treatment by castration (Figure 5, route 2).

Prostate cancer cells with an AR amplification could arise as small clonal growths in the tumour and be selected for by (1) androgen deprivation therapy or (2) natural androgen insufficiency within the tumour environment.
A critical question is, what proportion of castration-resistant patients that harbour an AR amplification could be attributable to pre-existing tumour clones in primary untreated tumours? Published data report that around 20% of primary tumours including those diagnosed by TURP (Edwards et al, 2003; Reid et al, 2010) progress to CRPCa (Byar, 1972; Taplin et al, 1995; Edwards et al, 2003; Cuzick et al, 2006), and 20–30% of these CRPCa harbour an AR amplification (Koivisto et al, 1997; Palmberg et al, 1997; Bubendorf et al, 1999; Brown et al, 2002; Edwards et al, 2003). Therefore, 4–6% of primary tumours are predicted to progress to CRPCa that contain a detectable AR amplification. Our studies have found an AR amplification in 6 of 596 patients (1%), which is a substantially lower rate of detection. However, this discrepancy could be explained by the small size of the AR-amplified clonal growths, which were less than 1% of tumour volume on whole section analysis, combined with the limited sampling that is an inherent problem of TMAs.
When taken together with the poor survival of AR-amplified patients, these data suggest that a significant proportion of AR-amplified CRPCa could develop from small clonal growths of AR-amplified cells present in pre-treatment neoplasms. Patients harbouring such clones would be predicted to be inherently more resistant to conventional androgen ablation therapy and would be excellent targets for first-line therapy with drugs that cause additional androgen ablation such as abiraterone (Visakorpi et al, 1995; Palmberg et al, 1997; Bonkhoff and Berges, 2010; De Bono and Ashworth, 2010) and/or the high-affinity anti-androgen enzalutamide (Bonkhoff and Berges, 2010; Scher et al, 2010). It is unlikely that such patients would be identified at a significant frequency using standard prostate biopsy procedures because of the small size of the AR-amplified clonal foci. However, as (i) PCa is identified as an incidental finding in ∼5% of men undergoing TURP as a treatment for BPH (Bubendorf et al, 1999; Melchior et al, 2009, ii) ∼20% of PCa occurs in the transition zone, with a significant number continuing to be diagnosed by TURP (Edwards et al, 2003; Berney, 2011) and (iii) treatment decisions for TURP-diagnosed PCa remain difficult (Cuzick et al, 2006; Berney, 2011), AR FISH mapping of such specimens could, in principle, provide a method of identifying men who should immediately receive more aggressive therapy.
References
Attard G, Clark J, Ambroisine L, Fisher G, Kovacs G, Flohr P, Berney D, Foster CS, Fletcher A, Gerald WL, Moller H, Reuter V, De Bono JS, Scardino P, Cuzick J, Cooper CS TransAtlantic Prostate Group (2008) Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene 27: 253–263.
Berney DM (2011) The tumour-node-metastasis staging of prostate cancer in transurethral resection of the prostate chips requires revision. BJU Int 107: 351–352.
Berney DM, Gopalan A, Kudahetti S, Fisher G, Ambroisine L, Foster CS, Reuter V, Eastham J, Moller H, Kattan MW, Gerald W, Cooper C, Scardino P, Cuzick J (2009) Ki-67 and outcome in clinically localised prostate cancer: analysis of conservatively treated prostate cancer patients from the Trans-Atlantic Prostate Group study. Br J Cancer 100: 888–893.
Bonkhoff H, Berges R (2010) From pathogenesis to prevention of castration resistant prostate cancer. Prostate 70: 100–112.
Brown RSD, Edwards J, Dogan A, Payne H, Harland SJ, Bartlett JMS, Masters JRW (2002) Amplification of the androgen receptor gene in bone metastases from hormone-refractory prostate cancer. J Pathol 198: 237–244.
Bubendorf L, Kononen J, Koivisto P, Schraml P, Moch H, Gasser TC, Willi N, Mihatsch MJ, Sauter G, Kallioniemi OP (1999) Survey of gene amplifications during prostate cancer progression by high-throughout fluorescence in situ hybridization on tissue microarrays. Cancer Res 59: 803–806.
Byar DP (1972) Survival of patients with incidentally found microscopic cancer of the prostate: results of a clinical trial of conservative treatment. J Urol 108: 908–913.
Clark J, Attard G, Jhavar S, Flohr P, Reid A, de Bono J, Eeles R, Scardino P, Cuzick J, Fisher G, Parker MD, Foster CS, Berney D, Kovacs G, Cooper CS (2008) Complex patterns of ETS gene alteration arise during cancer development in the human prostate. Oncogene 27: 1993–2003.
Cottu PH, Asselah J, Lae M, Pierga J-Y, Diéras V, Mignot L, Sigal-Zafrani B, Vincent-Salomon A (2008) Intratumoral heterogeneity of HER2/neu expression and its consequences for the management of advanced breast cancer. Ann Oncol 19: 595–597.
Cox D, Oakes D (1984) Analysis of Survival Data. Chapman & Hall: London. 201.
Cuzick J, Fisher G, Kattan MW, Berney D, Oliver T, Foster CS, Møller H, Reuter V, Fearn P, Eastham J, Scardino P Transatlantic Prostate Group (2006) Long-term outcome among men with conservatively treated localised prostate cancer. Br J Cancer 95: 1186–1194.
De Bono JS, Ashworth A (2010) Translating cancer research into targeted therapeutics. Nature 467: 543–549.
Edwards J, Krishna NS, Grigor KM, Bartlett JMS (2003) Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br J Cancer 89: 552–556.
Haapala K, Kuukasjärvi T, Hyytinen E, Rantala I, Helin HJ, Koivisto PA (2007) Androgen receptor amplification is associated with increased cell proliferation in prostate cancer. Hum Pathol 38: 474–478.
Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, Trapman J, Cleutjens K, Noordzij A, Visakorpi T, Kallioniemi OP (1997) Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res 57: 314–319.
Linja MJ, Savinainen KJ, Saramäki OR, Tammela TL, Vessella RL, Visakorpi T (2001) Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res 61: 3550–3555.
Melchior S, Hadaschik B, Thüroff S, Thomas C, Gillitzer R, Thüroff J (2009) Outcome of radical prostatectomy for incidental carcinoma of the prostate. BJU Int 103: 1478–1481.
Palmberg C, Koivisto P, Hyytinen E, Isola J, Visakorpi T, Kallioniemi OP, Tammela T (1997) Androgen receptor gene amplification in a recurrent prostate cancer after monotherapy with the nonsteroidal potent antiandrogen Casodex (bicalutamide) with a subsequent favorable response to maximal androgen blockade. Eur Urol 31: 216–219.
Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ (2008) Efficient tumour formation by single human melanoma cells. Nature 456: 593–598.
Reid AHM, Attard G, Ambroisine L, Fisher G, Kovacs G, Brewer D, Clark J, Flohr P, Edwards S, Berney DM, Foster CS, Fletcher A, Gerald WL, Moller H, Reuter VE, Scardino PT, Cuzick J, De Bono JS, Cooper CS Transatlantic Prostate Group (2010) Molecular characterisation of ERG, ETV1 and PTEN gene loci identifies patients at low and high risk of death from prostate cancer. Br J Cancer 102: 678–684.
Salonen AJ, Taari K, Ala-Opas M, Viitanen J, Lundstedt S, Tammela TLJ the FinnProstate Group (2013) Advanced Prostate Cancer Treated with Intermittent or Continuous Androgen Deprivation in the Randomised FinnProstate Study VII: Quality of Life and Adverse Effects. Eur Urol 63: 111–120.
Scher HI, Beer TM, Higano CS, Anand A, Taplin M-E, Efstathiou E, Rathkopf D, Shelkey J, Yu EY, Alumkal J, Hung D, Hirmand M, Seely L, Morris MJ, Danila DC, Humm J, Larson S, Fleisher M, Sawyers CL Prostate Cancer Foundation/Department of Defense Prostate Cancer Clinical Trials Consortium (2010) Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet 375: 1437–1446.
Shackleton M, Quintana E, Fearon ER, Morrison SJ (2009) Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell 138: 822–829.
Taplin M-E, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, Keer HN, Balk SP (1995) Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med 332: 1393–1398.
Taplin M-E, Rajeshkumar B, Halabi S, Werner CP, Woda BA, Picus J, Stadler W, Hayes DF, Kantoff PW, Vogelzang NJ, Small EJ Cancer and Leukemia Group B Study 9663 (2003) Androgen receptor mutations in androgen-independent prostate cancer: Cancer and Leukemia Group B Study 9663. J Clin Oncol 21: 2673–2678.
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS et al (2010) Integrative genomic profiling of human prostate cancer. Cancer Cell 18: 11–22.
Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinänen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP (1995) In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet 9: 401–406.
Waltering KK, Helenius MA, Sahu B, Manni V, Linja MJ, Jänne OA, Visakorpi T (2009) Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res 69: 8141–8149.
Waltering KK, Urbanucci A, Visakorpi T (2012) Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol Cell Endocrinol 360: 38–43.
Warburton PE, Greig GM, Haaf T, Willard HF (1991) PCR amplification of chromosome-specific alpha satellite DNA: definition of centromeric STS markers and polymorphic analysis. Genomics 11: 324–333.
Acknowledgements
This work was funded by the International Association of Cancer research, the Bob Champion Cancer Trust, the Grand Charity of Freemasons, the Big C Cancer Charity, Orchid and Prostate Cancer UK.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Merson, S., Yang, Z., Brewer, D. et al. Focal amplification of the androgen receptor gene in hormone-naive human prostate cancer. Br J Cancer 110, 1655–1662 (2014). https://doi.org/10.1038/bjc.2014.13
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.13
Keywords
This article is cited by
-
Acquired copy number variation in prostate tumours: a review of common somatic copy number alterations, how they are formed and their clinical utility
British Journal of Cancer (2024)
-
Androgen receptor variant-driven prostate cancer: clinical implications and therapeutic targeting
Prostate Cancer and Prostatic Diseases (2016)
-
Androgen Receptor Splice Variants in the Era of Enzalutamide and Abiraterone
Hormones and Cancer (2014)
