
Abstract
Background:
This study evaluated the addition of sorafenib to gemcitabine and cisplatin in biliary adenocarcinoma first-line therapy.
Methods:
Patients with advanced biliary adenocarcinomas received gemcitabine 1000 mg m−2 and cisplatin 25 mg m−2 on a 2 weeks on/1 week off cycle and sorafenib 400 mg twice daily. After the initial 16 patients were enrolled, the chemotherapy doses were amended in view of grade 3 and 4 hand–foot skin reaction and haematologic toxicity. Subsequently, 21 patients received gemcitabine 800 mg m−2, cisplatin 20 mg m−2 and sorafenib 400 mg. The primary end point was an improvement in 6-month progression-free survival (PFS6) from historical 57–77% (90% power, type I error of 10%). Pretreatment pERK, evaluated by immunostaining, was correlated with clinical outcome.
Results:
A total of 39 patients were accrued. The most common grade 3–4 toxicities noted in >10% of patients were fatigue, elevated liver function tests and haematologic toxicities including thromboemboli, hyponatraemia and hypophosphataemia. Six-month progression-free survival was 51% (95% confidence interval (CI) 34–66%). Median PFS and overall survival were 6.5 (95% CI: 3.5–8.3) and 14.4 months (95% CI: 11.6–19.2 months), respectively. No correlation was observed between pERK and outcomes.
Conclusion:
The addition of sorafenib to gemcitabine and cisplatin in biliary adenocarcinomas did not improve efficacy over historical data, and toxicity was increased.
Similar content being viewed by others

Main
Sorafenib is an oral antiangiogenic agent that targets VEGFR-2/3 and platelet-derived growth factor receptor-β and an inhibitor of the Raf/MEK/ERK signaling pathway at the level of Raf kinase (Wilhelm et al, 2004). The vascular endothelial growth factor receptor (VEGF) gene may be involved in biliary carcinomas (Benckert et al, 2003), with demonstrated VEGF overexpression in 53.8% and 59.2% of intrahepatic and extrahepatic cholangiocarcinomas, respectively (Yoshikawa et al, 2008). Activating BRAF gene mutations in the Raf/MEK/ERK pathway have been implicated in the development of biliary cancers and could represent a potential therapeutic target (Tannapfel et al, 2003). Cholangiocarcinoma cells are reported to become more susceptible to apoptosis with Raf-1 inhibitor BAY 37-9751, which is a close structural analogue of sorafenib, by blocking Mcl-1, an antiapoptotic protein (Yoon et al, 2002).
A current standard systemic therapy for advanced biliary adenocarcinomas is gemcitabine and cisplatin (Valle et al, 2010). The ABC-01 randomised phase II study evaluated the combination of gemcitabine and cisplatin and gemcitabine alone in patients with advanced biliary cancers (Valle et al, 2009). In an ‘exploratory’ comparison between the two arms, 6-month progression-free survival (PFS6) was 57.1% for the combination therapy vs 45.5% for single-agent gemcitabine. These results, in addition to an acceptable toxicity profile for the combination therapy, led to the expansion of the study into the ABC-02 trial, a randomised phase III study of 410 patients with advanced biliary tract cancers that demonstrated a median overall survival (OS) of 11.2 vs 7.7 months in favour of the gemcitabine plus cisplatin compared with gemcitabine monotherapy (P<0.001).
Given the preclinical evidence for potential clinical efficacy and the advent of gemcitabine plus cisplatin as a standard of care, a phase II study evaluating the combination of gemcitabine, cisplatin and sorafenib in patients with advanced biliary adenocarcinomas was undertaken.
Materials and methods
The study was a phase II, single-institution, non-randomised, open-label clinical trial in patients with advanced biliary adenocarcinomas. It was approved by the institutional review board at Memorial Sloan–Kettering Cancer Center. Written informed consent was obtained from each patient. The US National Cancer Institute ClinicalTrials.gov identifier is NCT00919061.
Eligibility criteria
Patients with histologically proven, non-resectable, recurrent or metastatic biliary adenocarcinoma including intrahepatic cholangiocarcinoma, extrahepatic cholangiocarcinoma and gallbladder adenocarcinoma with measurable disease by Response Evaluation Criteria in Solid Tumors (RECIST) 1.0 (Therasse et al, 2000) were eligible for enrolment. Patients with combined cholangiocarcinoma and hepatocellular carcinoma were also allowed in the study. No prior systemic therapy except for gemcitabine or 5-fluorouracil in the adjuvant setting, administered more than 6 months prior to enrolment, was permitted. Patients were required to have a Karnofsky performance status of ⩾80% and adequate organ function, including a haemoglobin level of ⩾8 g dl−1, ANC ⩾1.5 × 103 μl−1, platelet count ⩾100 × 103 μl−1, serum creatinine ⩽2 mg dl−1 or calculated creatinine clearance ⩾60 ml min−1, total bilirubin ⩽2 mg dl−1, and ALT and AST ⩽3 times the upper limit of normal (⩽5 times if liver metastases present). Patients with evidence of biliary obstruction were only allowed to join the study if total bilirubin level was expected to decrease to the required limit after adequate biliary drainage.
Treatment and dose modifications
Gemcitabine and cisplatin were administered as an intravenous infusion, weekly for 2 weeks, followed by a week off treatment for each 3-week cycle at starting doses of 1000 mg m−2 and 25 mg m−2, respectively. A maximum of 3-week delay in treatment was allowed. During the conduct of the study, in view of excess haematologic and other toxicity as described in the Results section, the study was amended and the gemcitabine dose was reduced to 800 mg m−2 and the cisplatin dose to 20 mg m−2 at the same treatment schedule. Treatment was continued until disease progression (either clinical or radiologic evidence) or development of unacceptable toxicity.
Sorafenib was prescribed at the standard dose of 400 mg twice daily throughout the cycle. Dose delays or modifications were required due to drug-related toxicities. A maximum of 3-week delay in treatment was allowed. During the conduct of the study, in view of unanticipated toxicity described in the Results section, the study was amended and sorafenib dose was reduced to 400 mg daily at the same treatment schedule.
Safety and tolerability
All patients were assessed for toxicity per the NCI Common Toxicity Criteria, version 3.
Tumour assessments
Restaging imaging studies were performed and levels of tumour markers CA 19-9 and CEA were obtained at baseline and subsequently after three cycles (every 9 weeks) of treatment. Treatment continued until there was evidence of disease progression, development of unacceptable toxicity or consent withdrawal. Thereafter, patients were continued to be followed up for survival. Tumour assessments were based on RECIST 1.0 guidelines.
Primary end point
The primary efficacy end point was the PFS6 rate. Progression-free survival (PFS) was calculated from study entry to documented disease progression or death from any cause, whichever occurred first. Patients who progressed at the 27-week planned scan, regardless of the actual calendar date of that scan, were declared as having progressed at 6 months. Patients who dropped out the study before 6 months due to toxicity without documented disease progression were counted as event for the primary end point. Patients who either withdrew consent prior to being treated or were unable to start treatment due to other complications were censored at 1 day after they consented on study.
Secondary end points
Median PFS, time to progression (TTP) and OS were determined. Time to progression was calculated from study entry to documented disease progression. Time to death was calculated from study entry to death or last follow-up. Disease-specific outcome subset analyses were performed.
Correlative studies
Pretreatment tissues were evaluated for pERK expression using rabbit polyclonal antibody phospho-p44/42 MAPK (Thr202/Tyr204) (Cell Signaling Technology Inc., Danvers, MA, USA). Positive and negative controls were used to ensure the integrity of all tumour samples and reagents. Stained slides were evaluated independently by a pathologist (JS). Localisation of pERK staining to cell nuclei or cytoplasm was evaluated qualitatively. Nuclear pERK staining intensity was graded semiquantitatively using a five-point scale: 0, no staining; 1+, weak; 2+, moderate; 3+, strong and 4+, intense. pERK expression was correlated with clinical outcomes, including PFS, response rate, TTP and OS.
Statistical design
Based on the ABC-01 and ABC-02 studies, PFS6 for the combination of gemcitabine and cisplatin are 57.1% and 59.3%, respectively (Valle et al, 2009). Using an exact single-stage binomial design with a desirable PFS6 of 77% and an undesirable PFS6 of 57%, the study called for enrolment of 39 patients to give a 90% power to detect the hypothesised improvement in the PFS6 rate for gemcitabine, cisplatin and sorafenib, with a type I error of 10%. The study regimen would be worthy of further investigation if 27 of the 39 patients were alive and progression free at 6 months. Progression-free survival, TTP and OS curves were estimated using the Kaplan–Meier methodology. The documented response rate and exact two-sided 95% confidence intervals (CIs) were calculated.
For correlative studies, PFS, TTP and OS were correlated with pERK staining using the univariate Cox regression model. Tumour response and pERK staining were correlated using Fisher’s exact test. All P-values were based on two-tailed statistical analysis. Statistical analysis was performed using SAS (version 9.2, Cary, NC, USA).
Results
Patient characteristics
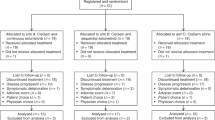
A total of 39 patients with advanced biliary tract carcinomas, with no prior systemic treatment, were enrolled in the study over a period of 16 months from 18 August 2009 to 28 December 2010. The analyses of the following results were performed on an intent-to-treat basis. Two patients were censored and were not evaluable for outcome: one patient withdrew consent from the study prior to being treated, and one patient was unable to start treatment due to complications from a small-bowel obstruction requiring surgical intervention. Among the enrolled patients, the median age was 65 years (range 31–83 years), with 20 patients (51%) being women. KPS ranged from 80 to 90%. Although the majority of patients (75%) were Caucasian, four (10%) patients were African-American and two (5%) patients were of Asian descent. In regard to the primary tumour site, 23 patients had intrahepatic cholangiocarcinoma (59%), with 2 patients (5%) had mixed intrahepatic cholangiocarcinoma plus hepatocellular carcinoma. Two patients (5%) had extrahepatic biliary tract carcinomas, and 14 patients (36%) had gallbladder adenocarcinoma. Notably, 38 patients (98%) had stage IV disease at time of study enrolment, whereas 1 patient had stage II disease.
Treatment
The median number of cycles of gemcitabine and cisplatin administered was 8 (range 1–27). The median doses of gemcitabine and cisplatin received were 800 mg m−2 (range 410–1000 mg m−2) and 20 mg m−2 (range 13–35 mg m−2), respectively. The median time of treatment on sorafenib was 5.6 months (range 0.2–21 months), with a median dose of 400 mg daily (range 200–800 mg).
Toxicity and dose modifications
After enrolment of the first 16 patients, the study was amended due to poor tolerability of the combination treatment with starting doses of gemcitabine 1000 mg m−2, cisplatin 25 mg m−2 and sorafenib 400 mg twice daily. The major adverse effects were grade 3 hand–foot syndrome in four (25%) patients and grade 3 or 4 haematologic toxicities. These included five patients (31%) with anaemia, five patients (31%) with grade 3 or 4 neutropenia and five (31%) patients with grade 3 or 4 thrombocytopenia. In addition, five patients were found to have a thrombotic or embolic event. After dose modifications, 21 newly enrolled patients were treated with gemcitabine 800 mg m−2, cisplatin 20 mg m−2 and sorafenib 400 mg once daily using the same treatment schedule. Twenty-one patients accrued subsequently received treatment at the modified starting doses.
The major adverse effects of the combination therapy throughout the study, including grade 3 and 4 toxicities noted in >10% of study patients, are listed in Table 1. The most common of which were fatigue, elevated liver function tests and haematologic toxicities including thromboemboli, hyponatraemia and hypophosphataemia.
Patient disposition
Of the 37 evaluable patients, 20 patients were discontinued from the study because of disease progression. An additional 15 patients came off the study due to treatment-related toxicities. These included two patients with myocardial infarctions, three with nephrotoxicity, three with neuropathy, three with thrombocytopenia, one with hyperbilirubinaemia, one with syndrome of inappropriate antidiuretic hormone, one with hand–foot skin reaction and one with fatigue. One patient subsequently underwent surgery, and one patient passed away while on study due to his cancer. Four additional patients died within 30 days of discontinuation of therapy: three due to progression of disease and one due to respiratory failure secondary to systemic infection and pulmonary embolism. A total of 26 patients (67%) received second-line or third-line therapies after disease progression or intolerable toxicities. The most common regimen used was FOLFIRI and single agents capecitabine and irinotecan.
Clinical outcomes
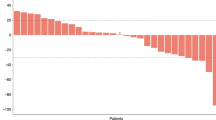
Progression-free survival at 6 months was 51% (95% CI, range 34–66%) (Figure 1). The median PFS was 6.5 months (95% CI: 3.5–8.3 months). Sensitivity analysis for patients who came off the study due to toxicities was performed with the same PFS results. The median TTP was 8.2 months (95% CI: 6.5–12 months). The median OS was 14.4 months (95% CI: 11.6–19.2 months) (Figure 2). There were no complete tumour responses reported. In total, 6 patients came off the study prior to undergoing restaging scans, and there were 33 evaluable patients for best response outcome: 4 patients (12%, 95% CI: 3–28%) had partial responses; 25 patients (76%, 95% CI: 58–88%) had stable disease, with median time in stable disease of 8.3 months (range 2.1–27.9 months); and 4 patients (12%, 95% CI: 3–28%) had disease progression.

Kaplan–Meier curve of PFS with treatment.

OS in patients who received gemcitabine, cisplatin and sorafenib therapy.
Disease-specific subset analyses were limited to gallbladder cancer and intrahepatic cholangiocarcinoma, in view of the very limited number of cases with extrahepatic cholangiocarcinoma and intrahepatic cholangiocarcinoma plus hepatocellular carcinoma. There was no association between OS and disease type (gallbladder cancer vs intrahepatic cholangiocarcinoma). P-value from the log-rank test was 0.71. There was also no association between PFS and disease type: P-value from the log-rank test was 0.22. Based on PFS sensitivity analysis, for patients came off the study because of toxicity, there was also no association between PFS and disease type: P-value from the log-rank test was 0.20. There was, however, a significant association between TTP and disease type. Specifically, median TTP for patients diagnosed with gallbladder disease was 5 months (95% CI: 2.3–8.9 months) compared with 10.4 months (95% CI: 6.4–14.5 months) for those who were diagnosed with intrahepatic bile duct cancer (P-value from the log-rank test was 0.013).
pERK expression
Thirty-one patients had tumour tissue available for pERK staining. In the majority of tumour samples, pERK staining was most intense within the nucleus of tumour cells. There was no expression noted in 10% of the cases, 1+ (weak staining) in 16% of samples, 2+ (moderate staining) in 29% of samples and 3+ (strong staining) in 45% of samples.
There was no correlation between pERK staining intensity and the clinical outcomes measured. Patients with tumour cells expressing higher pERK staining intensity had no improvement in their PFS, TTP or OS.
Discussion
The addition of sorafenib to gemcitabine and cisplatin combination therapy failed to show an improvement in PFS6 when compared with historical control data. The favourable median OS of 14.4 months is probably a consequence of a single institutional experience and relatively median KPS of 80% and age of 65 years. Of note, 72% went on to receive a second-line therapy (N=28, 72%). The latter observation may also be explained by the better selection of patients in the single-institution study compared with the ABC-02 phase III randomised study (Valle et al, 2010).
The combination therapy was administered at the standard starting doses based on the safety data from previous phase I studies of gemcitabine plus sorafenib and platinum plus sorafenib combination therapies (Kupsch et al, 2005; Siu et al, 2006). However, common practice suggests difficulty in maintaining these doses of cisplatin and gemcitabine for very long. Given the poor tolerability to the triple combination therapy at the standard starting doses, this study ultimately required dose reduction of all three agents after enrolment of 16 patients. This is a limitation of the reported herein study where a formal phase I study to evaluate the safety and tolerability of the gemcitabine, cisplatin and sorafenib combination would have been very valuable.
An unplanned analysis of patients with full vs reduced dose of the combination therapy showed no differences in the clinical outcomes measured attributable to dosing differences. The median duration of treatment was 24 weeks (eight cycles), which is comparable to the 21 weeks in the gemcitabine plus cisplatin arm in the ABC-02 study (Valle et al, 2010).
Notwithstanding the role of the Raf/MEK/ERK pathway in biliary tract cancers, there was no correlation between pretreatment tumour cell pERK expression levels and outcome. Comparison of pERK expression in sequential pretreatment and post-treatment biopsies in patients on therapy may provide important information on the tumour response to treatment. A study evaluating sorafenib at the full dose of 400 mg orally twice daily as a single agent in the first-line setting in patients with advanced biliary adenocarcinomas was ongoing at the time of the reported herein trial and was recently published (El-Khoueiry et al, 2012). There were no confirmed responses. Two patients (6%) achieved partial responses of short-lived duration. Ten (32%) patients had stable disease, with a median TTP of 4.4 months (95% CI: 3–7 months). The most common grade 3 and 4 toxicities were hand–foot skin reaction (13%), hyperbilirubinaemia (13%), venous thromboembolism (10%) and elevated liver function tests (10%). The study was terminated early. Recently and after initiation of the current study, sorafenib was identified to be an ineffective B-Raf inhibitor. In a phase II trial evaluating sorafenib in melanoma patients, BRAF (V600E) mutational status of the tumour was not associated with clinical activity and no significant effect of sorafenib on cyclin D1 or Ki67 was seen (Ott et al, 2010).
This study, like most biliary studies, included biliary adenocarcinomas of all subtypes regardless of the known differences in the pathology and molecular biology of the differing tumour subtypes. This point continues to be a subject of debate, especially in light of recent studies that show the variable expression of genes involved in cell cycle control and EGFR expression among biliary cancers from different sites of origin (Jarnagin et al, 2006; Yoshikawa et al, 2008). In addition, some studies suggest that response rates to cytotoxic therapies may be different, with higher response rates seen in patients with gallbladder carcinoma and extrahepatic cholangiocarcinomas than in patients with intrahepatic cholangiocarcinomas (Harder et al, 2006; Nehls et al, 2008). In this study, we noted a significant difference in TTP between gallbladder cancer and intrahepatic cholangiocarcinoma; however, this significant difference was not apparent when comparing PFS and OS. Although these findings support the need to differentiate and stratify patients with advanced biliary tract cancers by the sites of origin, they add the need to understand not only the molecular aspects of the different subtypes of biliary adenocarcinomas but other confounding variables, that is, clinical complications that may have effaced the difference noted in TTP when comparing PFS and OS, in the example herein.
In summary, the addition of sorafenib to gemcitabine and cisplatin in advanced biliary tract cancers failed to show any improvement in outcome and proved to have significant toxicity at standard cytotoxic dosing. The combination of gemcitabine, cisplatin and sorafenib is not recommended for further study in biliary cancers.
Change history
20 August 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Benckert C, Jonas S, Cramer T, Von Marschall Z, Schafer G, Peters M, Wagner K, Radke C, Wiedenmann B, Neuhaus P, Höcker M, Rosewicz S (2003) Transforming growth factor beta 1 stimulates vascular endothelial growth factor gene transcription in human cholangiocarcinoma cells. Cancer Res 63: 1083–1092.
El-Khoueiry AB, Rankin CJ, Ben-Josef E, Lenz HJ, Gold PJ, Hamilton RD, Govindarajan R, Eng C, Blanke CD (2012) SWOG 0514: a phase II study of sorafenib in patients with unresectable or metastatic gallbladder carcinoma and cholangiocarcinoma. Invest New Drugs 30: 1646–1651.
Harder J, Riecken B, Kummer O, Lohrmann C, Otto F, Usadel H, Geissler M, Opitz O, Henss H (2006) Outpatient chemotherapy with gemcitabine and oxaliplatin in patients with biliary tract cancer. Br J Cancer 95: 848–852.
Jarnagin WR, Klimstra DS, Hezel M, Gonen M, Fong Y, Roggin K, Cymes K, DeMatteo RP, D’Angelica M, Blumgart LH, Singh B (2006) Differential cell cycle-regulatory protein expression in biliary tract adenocarcinoma: correlation with anatomic site, pathologic variables, and clinical outcome. J Clin Oncol 24: 1152–1160.
Kupsch P, Henning BF, Passarge K, Richly H, Wiesemann K, Hilger RA, Scheulen ME, Christensen O, Brendel E, Schwartz B, Hofstra E, Voigtmann R, Seeber S, Strumberg D (2005) Results of a phase I trial of sorafenib (BAY 43-9006) in combination with oxaliplatin in patients with refractory solid tumors, including colorectal cancer. Clin Colorectal Cancer 5: 188–196.
Nehls O, Oettle H, Hartmann JT, Hofheinz RD, Hass HG, Horger MS, Koppenhöfer U, Hochhaus A, Stieler J, Trojan J, Gregor M, Klump B (2008) Capecitabine plus oxaliplatin as first-line treatment in patients with advanced biliary system adenocarcinoma: a prospective multicentre phase II trial. Br J Cancer 98: 309–315.
Ott PA, Hamilton A, Min C, Safarzadeh-Amiri S, Goldberg L, Yoon J, Yee H, Buckley M, Christos PJ, Wright JJ, Polsky D, Osman I, Liebes L, Pavlick AC (2010) A phase II trial of sorafenib in metastatic melanoma with tissue correlates. PLoS One 5: e15588.
Siu LL, Awada A, Takimoto CH, Piccart M, Schwartz B, Giannaris T, Lathia C, Petrenciuc O, Moore MJ (2006) Phase I trial of sorafenib and gemcitabine in advanced solid tumors with an expanded cohort in advanced pancreatic cancer. Clin Cancer Res 12: 144–151.
Tannapfel A, Sommerer F, Benicke M, Katalinic A, Uhlmann D, Witzigmann H, Hauss J, Wittekind C (2003) Mutations of the BRAF gene in cholangiocarcinoma but not in hepatocellular carcinoma. Gut 52: 706–712.
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92: 205–216.
Valle JW, Wasan H, Johnson P, Jones E, Dixon L, Swindell R, Baka S, Maraveyas A, Corrie P, Falk S, Gollins S, Lofts F, Evans L, Meyer T, Anthoney A, Iveson T, Highley M, Osborne R, Bridgewater J (2009) Gemcitabine alone or in combination with cisplatin in patients with advanced or metastatic cholangiocarcinoma or other biliary tract tumours: a multicentre randomized phase II study – the UK ABC-01 study. Br J Cancer 101: 621–627.
Valle JW, Wasan HS, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, Madhusudan S, Iveson T, Hughes S, Pereira SP, Roughton M, Bridgewater J ABC-02 Trial Investigators (2010) Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 362: 1273–1281.
Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA (2004) BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 64: 7099–7109.
Yoon JH, Werneburg NW, Higuchi H, Canbay AE, Kaufmann SH, Akgul C, Edwards SW, Gores GJ (2002) Bile acids inhibit Mcl-1 protein turnover via an epidermal growth factor receptor/Raf-1-dependent mechanism. Cancer Res 62: 6500–6505.
Yoshikawa D, Ojima H, Iwasaki M, Hiraoka N, Kosuge T, Kasai S, Hirohashi S, Shibata T (2008) Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br J Cancer 98: 418–425.
Acknowledgements
This study was generously supported by a research grant from Bayer Pharmaceuticals. The sponsor’s role was limited to research funding and a courtesy review of the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Marinela Capanu, Eileen M O’Reilly, Jennifer Ma, Joanne F Chou, Bolorsukh Gansukh, Leonard B Saltz and Ghassan K Abou-Alfa have received research grant support. Eileen M O’Reilly and Ghassan K Abou-Alfa were consultants.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Lee, J., Capanu, M., O'Reilly, E. et al. A phase II study of gemcitabine and cisplatin plus sorafenib in patients with advanced biliary adenocarcinomas. Br J Cancer 109, 915–919 (2013). https://doi.org/10.1038/bjc.2013.432
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.432
Keywords
This article is cited by
-
Systemic treatment options for advanced biliary tract carcinoma
Journal of Gastroenterology (2020)
-
α2,6-Sialylation promotes immune escape in hepatocarcinoma cells by regulating T cell functions and CD147/MMP signaling
Journal of Physiology and Biochemistry (2019)
-
Efficacy and safety of chemotherapy with or without targeted therapy in biliary tract cancer: A meta-analysis of 7 randomized controlled trials
Journal of Huazhong University of Science and Technology [Medical Sciences] (2017)
-
A Phase I, Dose-Escalation Trial of Pazopanib in Combination with Cisplatin in Patients with Advanced Solid Tumors: A UNICANCER Study
Oncology and Therapy (2016)
-
Genomics of gallbladder cancer: the case for biomarker-driven clinical trial design
Cancer and Metastasis Reviews (2016)
