
Abstract
Next-generation sequencing (NGS) of cancer genomes promises to revolutionise oncology, with the ability to design and use targeted drugs, to predict outcome and response, and to classify tumours. It is continually becoming cheaper, faster and more reliable, with the capability to identify rare yet clinically important somatic mutations. Technical challenges include sequencing samples of low quality and/or quantity, reliable identification of structural and copy number variation, and assessment of intratumour heterogeneity. Once these problems are overcome, the use of the data to guide clinical decision making is not straightforward, and there is a risk of premature use of molecular changes to guide patient management in the absence of supporting evidence. Paradoxically, NGS may simply move the bottleneck of personalised medicine from data acquisition to the identification of reliable biomarkers. Standardised cancer NGS data collection on an international scale would be a significant step towards optimising patient care.
Similar content being viewed by others

Main
Molecular diagnostics has a key role in medicine in diagnosing and classifying diseases, and increasingly in tailoring treatments to individuals. Examples of personalised cancer medicine have emerged in recent years with the use of somatic mutations in EGFR and BRAF to determine treatment response, predict survival and direct the selection of patients for treatment with gefitinib and vemurafenib, respectively (Mok et al, 2009; Chapman et al, 2011). Until recently, small-scale methods such as classical (Sanger) sequencing or pyrosequencing were used to identify mutations in genes such as these. Anticipated developments in molecularly targeted therapies mean that it may be necessary to sequence large panels of genes in cancers to allow the best therapies to be chosen. In addition, reliable DNA-based molecular markers of prognosis are gradually being identified, and these can be used to modulate treatment, adding further to the demand for large-scale DNA sequencing.
In recent years, next-generation sequencing (NGS) has begun to supplant other technologies. It can analyse entire human genomes in days and can produce sequences at a sub-genomic level in a clinically useful time frame. Progress in NGS has accelerated owing to several factors. Advances in nanotechnology have improved sequencing methods and this has been coupled with continued developments in bioinformatics. Faster and cheaper generation of data has been accompanied by increased accuracy to a level that emulates 1G (first generation – Sanger) sequencing and sufficient for clinical application.
To date, molecular diagnostics has largely used NGS to analyse patients’ constitutional DNA – to test for disease susceptibility mutations or drug toxicity variants – or to sequence pathogen genomes. By comparison, large-scale sequencing of cancer genomes requires another level of analytical complexity: the variety of complex genetic aberrations found in cancers means that it is not always possible to rely on standard human reference sequences, genes may be present in multiple copies, chromosome-scale (structural) changes are frequent, epigenomic changes are common, and intratumour genetic heterogeneity is likely to be present (Stratton et al, 2009). In short, accurate ascertainment of biomarkers and actionable drug targets in cancer genomes remains a considerable challenge. The objective of this review is to discuss the principles underlying the DNA-based analysis of cancer genomes using NGS, to present the state of the art and to consider critically how it could be used in clinical practice in the near future.
NGS platforms: technical aspects
The pioneering so-called 2G NGS platforms available at the time of writing are based on sequencing, in parallel, vast numbers of genome fragments sheared into lengths of approximately 35–400 bp. These fragments are clonally amplified by emulsion polymerase chain reaction (emPCR; Life Technologies, Applied Biosystems SOLiD, Carlsbad, CA, USA; Roche 454, Branford, CT, USA), by immobilisation on a solid surface (Illumina, San Diego, CA, USA) or by nanoball amplification (Complete Genomics, Mountain View, CA, USA). In all cases, fluorescence emission from a nucleotide added in a sequencing synthesis reaction, either directly or by use of a probe, is captured to determine the genomic sequence. The leading 2G platforms using this technology include the Illumina genome analyser, which is currently the dominant platform in the field, the Applied Biosystems SOLiD platform, the Complete Genomics and the Roche Applied Science 454 genome sequencer (Metzker, 2010). While based on similar concepts, the particular features of each platform lead to specific characteristics in terms of speed, length of sequenced fragments (reads) and accuracy. In general, longer reads are desirable, especially where mapping the location of a sequence is problematic and mutations other than base substitutions are expected, but there appears to be a trade-off between read length and factors such as cost and, arguably, accuracy.
With Illumina sequencing technology, genomic DNA is sheared to form fragments of 75–150 bp. Adapters are ligated to the fragments and bind to the surface of a flow cell channel. The fragments are then amplified using bridge amplification. Following this, sequencing commences by adding four labelled reversible terminating nucleotides, DNA polymerase and primers. Upon addition of the terminating nucleotide, a fluorescent signal is emitted that is captured and denotes the type of base incorporated in the sequence. The sequencing cycle is repeated one base at a time generating a series of images with each image representing a single base in the priming sequence.
With the ABI SOLiD platform, DNA fragments are bound to clonal bead populations. The fragments are attached to the beads with a universal adaptor sequence. Emulsion polymerase chain reaction occurs in microreactors and the products from the PCR, which are attached to the beads, are bound to a glass slide. Primers hybridise to the adaptor sequence and fluorescently labelled di-base probes are added and compete to ligate to the priming sequence. There is increased specificity with this method and SOLiD produces a signal per one pair of bases, with read lengths of up to 75 bp.
In the Roche 454 system, DNA fragments are attached to beads and undergo bead immobilised clonal amplification. The beads are exposed to the four DNA nucleotides sequentially with nucleotides being incorporated when they complement the template strand. The incorporation of nucleotide results in the emission of a light signal, which is proportional to the number of nucleotides. The Roche 454 has much longer reads (400 bp), but signal intensity falls in stretches of identical consecutive bases.
Complete genomics are currently available only on a service basis and uses an unusual technology to circularise DNA fragment within a vector. This results in relatively short (∼35 bp), gapped reads.
Newer NGS platforms (3G) utilise single molecule templates, which do not require PCR amplification. The benefits of this are two-fold. They require less starting material and sequencing a single template is less error prone, since it avoids the introduction of sequencing errors, which could otherwise occur through clonal amplification from PCR. Lead developers in the single template technology include Helicos BioSciences (Cambridge, MA, USA) and Pacific BioSciences (Menlo Park, CA, USA). Such is the rate of progress that 4G sequencers are on the horizon. An example is the use of single molecule sequencing incorporating nanopore technology (Venkatesan and Bashir, 2011). Oxford Nanopore Technologies (Oxford, UK) has recently announced the intended production of a USB-size portable DNA sequencer.
The main NGS platforms are also suitable for targeted genome-wide (but not whole-genome) sequencing. Frequently, the targets comprise the 1% of DNA that encodes mRNA (‘the exome’) and enrichment for these sequences can be performed using oligonucleotide-based capture methods of genomic DNA in solution. Proprietary methods are available from companies such as Agilent (Santa Clara, CA, USA) and Illumina provide standard exome panel capture kits. Increasingly, other targeted capture panels are being made available and custom panels can be designed by users. However, in all cases, capture efficiency is inevitably variable across the genome regions targeted (Parla et al, 2011) and, furthermore, coverage of each targeted region needs to increase progressively from zero on the outer boundaries to the expected depth, resulting in a need for greater median depth (100X or more) compared with whole-genome approaches. Whole-exome approaches currently tend to be used in research screens where there is a focus on coding variation. As sequencing costs fall, they may be supplanted by whole-genome NGS.
The quantity of DNA required for whole-genome sequencing or whole-exome sequencing varies dependent on the platform provider. The limiting factor is the amount of DNA required to produce an optimal library for sequencing. However, sequencing is now routinely performed on 100 ng DNA or less, and single-cell sequencing has been undertaken in several laboratories. Similarly, the cost for sequencing varies greatly among platform providers but whole-exome sequencing can typically be 10-fold less costly than whole-genome sequencing.
The drive to produce increasingly compact, cost-effective and time-efficient sequencing platforms has also resulted in products that can be utilised more readily for targeted sequencing of genes with easier sample preparation protocols, shorter run times and simpler data analysis (Desai and Jere, 2012). Some distributors have therefore developed a dual strategy to target the low-throughput clinical and high-throughput research environments (Illumina with MiSeq and HiSeq, respectively, and Roche with the GS Junior and FLX systems). The sequencing machines designed for immediate clinical applications are considerably cheaper. They aim to have faster run times providing less data while retaining similar qualities. The underlying sequencing technology is based on principles similar to their parent machines, which were originally designed for research purposes. The Miseq machine developed by Illumina reportedly has a run time as short as 8 h from the preparation of DNA to variant detection. In addition, it has the ability to run numerous samples from different patients simultaneously by tagging the samples with a barcode so that they can be identified. The Roche GS junior has an instrument run time of 10 h and has read lengths of 400 bp.
An innovative form of technology has been utilised by Life Technologies to produce bench top sequencers aimed for direct clinical applications. The platform uses semiconductors to determine the genomic sequence. In brief, the release of hydrogen ions on incorporation of a nucleotide into a DNA template has been exploited to determine the sequence of a piece of DNA (Rothberg et al, 2011). This method has the potential to enable cheaper and more rapid sequencing protocols than those relying on optical methods of detection. Life Sciences’ Ion Proton and Ion PGM utilise this method. The Life Sciences platforms require custom amplification of target sequences – typically tens of hundreds – before sequencing and special panels (e.g., of cancer genes) have been designed for this purpose. Illumina have recently also launched a custom amplification method (TSCA) that is designed principally for the MiSeq platform. In all custom amplification panels, special efforts must be made to equalise the efficiency of amplicon production across the panel, and the costs per base sequenced are generally higher than for capture methods.
In general, the choice of sequencer for clinical use will depend on the best combination of cost, flexibility, error rates, throughput and post-sequencing analysis programs for a given application. A comprehensive technical review of the main NGS platforms is given by Metzker (2010). In most cases where a sequencer is dedicated to clinical testing, smaller-scale machines have proved the most cost-effective and reliable.
Processing and analysis of NGS data
Each fragment of DNA is sequenced multiple times. The fragments can be sequenced from one end or from both ends matched to each other (‘paired ends’). Longer fragments of several kb can also be sequenced, often with the ends matched (‘mate pairs’). Paired-end reads can be used to identify small-scale genomic rearrangements, such as insertions and deletions, and they aid mapping of repetitive regions of the genome. Mate pair libraries are constructed to aid de novo assembly of genomes. Mate pair reads are typically sufficiently far apart (>5 kb) to provide a scaffold when reconstructing genomes and are potentially useful for identifying large structural variants.
The term ‘coverage’ (or ‘read depth’) is a reflection of how often a specific region of the genome has been sequenced. As perfect duplicate reads can contain errors arising from the amplification process, only unique reads with slightly different lengths or start/end points are analysed. After elimination of duplicate reads, most technologies aim for a median coverage in constitutional DNA of 30-fold (30X) at >90% of bases. The requirement for this sequencing depth is in order to allow proper mapping and assembly, and to differentiate between errors and true variants.
The reads resulting from sequencing are usually mapped by ‘comparative assembly’ to a reference genome such as the latest human whole-genome reference. This can be challenging, for example, owing to the presence of repeated sequences within the genome or large-scale polymorphisms. When selecting one of the several NGS mapping programs, one has to consider several factors. These include accuracy (proportion of reads mapped correctly), sensitivity (the proportion of reads mapped to the reference genome), time efficiency and the computing capacity required (Bao et al, 2011). The size of the genome being sequenced and the computing power available to the investigator will influence the decision in selecting a software package. The principles underlying mapping of reads to a reference genome are based on either indexing the genome or the reads themselves. Using such indexes, the genome can be mapped using computational algorithms. Two main approaches have been developed to map the genome, one involving a hash table-based algorithm and the other using a trie/Burrows–Wheeler transformation (Horner et al, 2010). The Burrows–Wheeler method relies on successive simple sorting transformations to better organise long strings of characters. This results in a single, reversible, permutation to make a complex string much easier to compress thanks to the colocation of repetitive characters. In genomics, the four-letter alphabet and the large number of repeats make this an ideal approach to handle the very large number of reads with a prefix tree structure, or trie. A key advantage of trie/Burrows–Wheeler algorithm-based software programs is their relatively low memory requirement.
The information generated from sequencing is stored in the form of a FASTQ file. This contains information regarding the sequence in each read and the quality of each base. In the process of mapping, a BAM file is created in which reads have been assigned a position relative to the reference genome while retaining information regarding unmapped reads. Examples of mapping programs include BWA (Li and Durbin 2009a, 2010) and Stampy (Lunter and Goodson, 2011). BWA utilises the Burrows–Wheeler transformation and is composed of three different algorithms. The choice of algorithm that should be utilised for a particular project is dependent on the read length and sensitivity required. Stampy utilises a hash-based algorithm to map Illumina reads to a reference genome with high sensitivity.
Following the mapping of the reads to a reference genome, putative mutations are identified by ‘variant calling’ programs. Sometimes, particular callers work better with particular mappers. Several statistical programs have been developed to call single nucleotide variants (SNVs), mostly for primary use in identifying germline variation in constitutional DNA. Commonly used programs include SAMtools (Li et al, 2009b), GATK (www.broadinstitute.org/gsa/wiki/index.php/Home_Page) and Platypus (www.well.ox.ac.uk/platypus). Information regarding the location of variant calls is generated in VCF files. Variant calls can be visualised in the VCF files and original BAM files using a visual tool display such as IGV (ThorvaldsdÓttir et al, 2013). ANNOVAR is a software engine, which annotates and filters variant calls (Wang et al, 2010). Using ANNOVAR variant call coordinates are assigned to a gene and filtered with regards to their frequencies in population databases to exclude germline non-pathogenic variations. The filtering process is necessary when trying to identify clinically relevant mutations.
Although there are several software programs available to map the genome and call variants, there is a lack of consensus on which tools are the best. The uncertainty underlying this stems from three reasons. First, programs are continually evolving, so that the existing programs rapidly become out-dated and therefore comparisons are made between asynchronous versions. Second, programs may be designed for selected sequencing platforms and their specific characteristics, and may therefore lack the capacity to process data from competing manufacturers; indeed, some programs from commercial sequencing vendors are not made freely available and the quality of analysis has to be taken ‘on trust’. Third, many mappers and callers perform poorly for mutations other than SNVs, and specialist programs are available to identify relatively uncommon types of mutations. Finally, there are limited data for direct comparisons of performances between the software programs.
Issues associated with the interpretation of NGS data
Conventional Sanger sequencing is perhaps no longer the gold standard for accurately sequencing small segments of genome. It is probably more error-prone than NGS, yet is still used to validate findings from NGS on the basis that the systematic errors associated with NGS and Sanger sequencing are different. Although NGS platforms have an apparently high accuracy, the sheer quantity of data means that getting 0.01% of the human genome wrong would correspond to 300 000 errors scattered along the 3 billion base pairs. It is only by identifying systematic errors for a fixed technology that one can avoid being overwhelmed by false-positive calls. In conventional Sanger sequencing, the accuracy of the call of each base in the sequence can be assessed using Phred scores, which are probability-based confidence scores (Ewing et al, 1998a; Ewing and Green, 1998b). Next-generation sequencing platforms have devised their own quality metric of ‘Phred-like’ scores to determine the accuracy of calling bases in the sequence. Notably, these quality scores relate to individual platforms, with difficulty in finding a consensus on how to measure accuracy across platforms. Typically the error rate from the pairs of base measured by SOLiD technology cannot be easily compared with the intensity-based 454, although characterisation of the matches and mismatches to some references does allow for some comparison. Technical errors in NGS tend to occur towards the ends of reads. Moreover, certain genomic features, such as GC-rich regions are difficult to sequence with most chemistries, while stretches of single base repeats are especially susceptible to artefactual insertion–deletion events with the current 454 technology.
Incorrect mapping of reads to the reference genome can be a further source of error. In theory, a random 30-bp read is of sufficient length to form a unique combination of the four-letter genetic code to map to a single location even in large genomes such as the 3 billion base pair long human genome (Horner et al, 2010). Despite using comparative assembly, a cohort of reads will remain either unmappable or incorrectly mapped. Variant calling may also fail, for example, if variant reads are mapped to a different location from reference reads or simply discarded as ‘unmappable’. Moreover, the thresholds used to call a variant vary from program-to-program, and user-to-user in terms of the total read depth required, consistency between sequencing reads on the two DNA strands, and the proportion of mutant reads in the total sequence required needed to assign a read as variant.
Cancer-specific considerations in NGS and data analysis
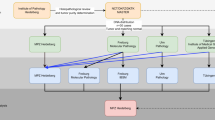
Next-generation sequencing has been utilised to characterise numerous cancers and large-scale projects such as The Cancer Genome Atlas (http://cancergenome.nih.gov/) and International Cancer Genome Consortium (http://icgc.org) now exist to comprehensively profile hundreds of cancers of any type using genome–exome sequencing, gene and protein expression profiling, RNA sequencing, methylome analysis, and copy number assessment. However, unlike most constitutional genomes, cancers are biologically diverse, heterogeneous entities with complex genetic alterations. Figure 1 summarises the process of analysing NGS data from cancer genomes. Below, we detail some of the problems and opportunities specific to cancer NGS.

Framework for cancer genome analysis using NGS.
Poor quality samples and limited DNA
The first consideration in cancer NGS is that almost all excised tumour samples have some degree of contamination from genetically normal tissue, although this can be minimised by using microdissection if necessary. However, many groups sequence cancers to higher median read depth (e.g., ∼50X for whole genomes) than that used for constitutional DNA. Lower depths of sequencing may be sufficient for tumours with a highly pure, diploid genome, such as some haematological malignancies. Close collaboration with histopathologists is necessary to ensure that the specimens are representative of tumour and to assess the degree of contamination from genetically normal tissue. Furthermore, many tumours take the form of formalin-fixed, paraffin-embedded (FFPE) material that consists of fragmented or cross-linked DNA that may have been further damaged over time (Gilbert et al, 2007). In addition, the quantity of DNA available may be too low for capture techniques generally used for exome sequencing. Hence, much of the NGS data on FFPE tissue have been derived from sequencing targeted amplicons (Kerick et al, 2011). Few successful genome or exome sequences from FFPE cancers have been reported to date, although we envisage improvements in this area.
Paired tumour–normal comparisons
In general, paired constitutional DNA from the patient’s normal tissue (often blood) is used to identify germline mutations, to differentiate between germline and somatic variants, to make analysis simpler and to provide some increase in error control. Following extraction of the DNA, the tumour and the normal samples are processed in parallel. As the use of normal comparators increases costs, some groups have dispensed with this for the analysis of specific, well-characterised variants that can be reliably detected and are highly unlikely ever to be present in the germline; examples include ‘hotspot’ mutations in genes such as KRAS and BRAF. The best source of constitutional DNA remains an issue. Many groups use peripheral blood, and this is generally expected to be acceptable. Matched normal tissue may be preferable if somatic mosaicism is a possibility. Too few data exist to allow the potential benefits of FFPE normal tissue alongside FFPE tumour to be assessed. The bioinformatic assessment of paired tumour–normal samples also continues to present a challenge. In brief, it may be necessary to use different variant calling thresholds to take account of factors that vary between the tumour and its paired normal sample, including read depth and aneuploidy/polyploidy.
Intratumour clonal heterogeneity
In addition to the fact that many regions of the cancer genome have a copy number that deviates from two, NGS has highlighted the fact that cancers often do not comprise a single dominant clone, but may have multiple subclones at non-trivial frequencies that comprise part of the entire tumour. It is not clear whether the subclones are important for tumour growth, but they may contain clinically important changes, such as mutations that confer resistance to chemotherapy. Recently, Gerlinger et al (2012) used exome sequencing in renal cancer to show the subclonal complexity and parallel evolution that can occur even in different parts of a primary carcinoma. This finding has significant clinical implications. For example, a single biopsy may not be representative of the genetic composition of the cancer, leading to inappropriate choice of treatment; and if a rare subclone exists, what is its importance for therapy? Intratumour heterogeneity is a formidable challenge that must be addressed in the development of personalised medicine and biomarker development.
Other cancer-specific factors
Many cancer genomes are highly rearranged, with multiple, large-scale mutations such as translocations, inversions, fusions and copy number changes. These factors make NGS analysis of tumours even more difficult. Some solutions are discussed further in the section below. Numerous other cancer-specific factors must be considered when undertaking NGS of all but the simplest, most-specific mutations. These include the presence of mutational signatures – including specific genetic defects in DNA repair and environmental exposures – that are often associated with a high background level of mutations. Non-human genomes sequences may also be present in tumour, for example, from oncogenic viruses or infections.
Working towards cancer-specific NGS solutions
Most bioinformatic tools for processing NGS have been designed for ‘normal’ genomes and the assumptions behind their application are commonly invalid in tumour samples for the reasons outlined in the previous section. However, there is often no warning of these limitations when such tools are misused and an output, however inaccurate, is often generated, so any assembly resulting for such non-cancer tools should be taken with caution. Analysis of cancer genomes therefore requires close collaboration between clinicians and bioinformaticians to optimally process data elicited from NGS to ensure that the data generated is clinically applicable.
More recently, cancer-specific tools have been developed to process NGS data (Table 1). Variant calling programs have been developed to call tumour and normal samples simultaneously so that only reads that are present in both files with a minimum coverage are called. Cancer-specific callers include JointSNVMix, Somatic Sniper, MuTect and Varscan2 (Koboldt et al, 2012; Larson et al, 2012; Roth et al, 2012; Cibulskis et al, 2013), were developed to identify somatic mutations from tumour-normal pairs. Varscan 2 has the ability to directly identify somatic indels and copy number variants (CNVs).
The identification of indel mutations of even a few base pairs is potentially very challenging, although this is often primarily a mapping rather than calling problem. Furthermore, larger indels of tens to thousands of bases, which are common in cancers, are unlikely to be encompassed in a single read and need special methods for their identification. One solution is the use of paired-end reads in sequencing. Here, a paired set of reads a known distance apart are sequenced and if the separation of the reads differs from that expected, an insertion or deletion may be the cause. This technique may also identify fusion genes, inversions and translocations. Several programs (e.g., BreakDancer, Dindel and Pindel) are available to identify indels (Chen et al, 2009; Ye et al, 2009; Albers et al, 2011). Their sensitivity and specificity are probably suboptimal, with quite different outputs from different programs and very high level of false-positive calls. One approach is to use several software programs to identify potential genetic aberrations and to validate these using other methods.
The roles of copy number variant CNVs and loss of heterozygosity in tumourigenesis are well established, for example, in the form of oncogene amplification. At present, there is a need for reliable tools to identify CNVs in cancer NGS data, although rapid developments in this field are anticipated. Two types of approaches can be taken and a combination of these may provide improved accuracy and allow a wide range of CNV sizes to be covered. First, CNVs can be regarded as very large indels of up to several megabases. This shows how difficult their identification can be, as they are beyond the reach of paired-end reads. Three methods are following this concept by either looking at recurrent splits within single reads, misalignment of paired reads or overall difficulty in mapping at a given location. Software such as Genome STRiP (Handsaker et al, 2011) extends the indel approach of paired reads and recurrent breakpoints to attempt to identify larger events, but these currently suffer from poor specificity and its focus is on common deletions across populations, with only limited applicability to cancer. The second approach is to use the number of reads at any site as an indicator of copy number. Analogous methods have already been developed for SNP array technology. OncoSNP, for example, uses a Bayes Hidden Markov Model and relies on allele-specific signal intensity (equivalent to read number) at polymorphic sites in comparison with other regions of the tumour genome and with the paired normal genome (Yau et al, 2010). It characterises copy number, loss of heterozygosity, intratumour heterogeneity and the degree of contamination by normal cells. Extension of such methods to NGS data is successfully under way, although exact performance is naturally uncertain at this point.
The discovery that some cancer chromosomes appear to have been smashed up and put back together (chromothrypsis) highlights just how complex their genomes can be. In some cases, therefore, de novo genome assembly may be required to fully identify the somatic mutation complement of any cancer. Programs such as SOAPdenovo2 (Luo et al, 2012) and ABySS (Birol et al, 2009) can be used here, especially if a genome scaffold is available from mate pair sequencing libraries. Eventually, such analysis may become routine, but we envisage that it will be confined to specialist research areas, rather than clinical practice, for the near future.
What sorts of results are found from cancer sequencing screens?
Although some cancer genomes probably contain over a million mutations at sufficient frequency to be identified by standard NGS methods, most have tens of thousands of base substitution and small indel mutations. The Cancer Genome Atlas Network (TCGAN) is actively characterising the genomic and epigenomic mutation spectrum in a variety of cancers. Although work in this field is ongoing we have some insight to what has been discovered from preliminary findings. Characterisation of squamous cell lung carcinomas, demonstrated that putative driver pathways involved in the initiation or progression of tumour development appear to have roles in oxidative stress and squamous differentiation (The Cancer Genome Atlas Research Network, 2012a). Squamous cell lung cancer appears to share many alterations that are in common with head and neck carcinomas without evidence of human papilloma virus infection involving genes such as TP53, CDKN2A, NOTCH1 and HRAS. This may suggest that the biology of the two diseases may be similar. Analysis of the genomic profile from exome sequencing in colorectal cancer demonstrated variations in the frequency of mutations in colorectal tumours (The Cancer Genome Atlas Research Network, 2012b). Hypermutation was associated with either POLE mutations or high levels of microsatellite instability because of hypermethylation and MLH1 silencing or somatic mutations in mismatch repair genes. A comprehensive analysis of breast tumours (The Cancer Genome Atlas Research Network, 2012c) demonstrated the existence of four main breast cancer classes defined by differing genomic and epigenetic abnormalities. Heterogeneity exists among breast cancers with only three genes prevalent at >10%, namely TP53, PIK3CA and GATA3 across all breast cancers. The mutation spectrum in basal-like breast cancers exhibited similarities with patients with serous ovarian carcinomas with TP53, RB1 and BRCA1 mutations and MYC amplification. This suggests a shared driving mechanism for tumour development and suggests that common therapeutics strategies could be considered.
The application of NGS in cancer molecular diagnostics
We have shown that many difficulties exist in the analysis of cancer NGS data, but NGS still promises to provide a more accurate picture of cancer as a somatic genetic disease than any other method used to date. The application of NGS in clinical and service laboratory practice is in its early stages, and the best way for this to proceed is unclear. One view is that the technology has such potential that its eventual introduction on a large scale is inevitable – whole-genome NGS should be introduced as soon as possible and we can wait for the number of useful tests to catch up with the technology. The opposing view is that we still have very few clinically useful tests available for any cancer type, and that focussed NGS should be introduced, and then only where there is clear superiority to other methods. As described above, NGS systems exist to cover both of these possibilities. At present, most diagnostic cancer NGS is of a targeted type, for example, covering the exomes of a few hundred genes. It is likely that the immediate application of NGS in the health-care service will focus on a targeted approach. The reducing sequencing costs and the capacity to sequence targeted genes in multiple patients concurrently will increase access to genetic testing and facilitate the development of personalised medicine. Table 2 demonstrates some of the potential clinical and research applications of NGS.
Familial genetic testing of cancers will benefit from NGS. Constraints from expenses and available resources associated with genetic testing from traditional sequencing limits the number of patients that are currently tested. Patients often have to meet stringent criteria based on their personal and family history before they are deemed eligible candidates for genetic testing. Numerous examples of familial genetic cancers exist, including BRCA1 and BRCA2 mutations in breast and ovarian cancer patients and APC in colorectal cancer. Møller et al (2007) reported that family history criteria detect <50% of the BRCA mutation carriers. Characterisation of the BRCA mutation in a cost-effective method could ensure that a larger number of patients are eligible for trials with poly (ADP-ribose) polymerase inhibitors.
In the era of personalised medicine, the use of targeted screening of selected genes will act as an invaluable tool to recruit patients to clinical studies. It is foreseeable that NGS technology will be used to recruit patients into clinical trials using molecularly targeted drugs. A high throughput of patients could undergo genetic testing to assess their eligibility to enrol in clinical trials within a clinically viable timeframe. For example, patients can be screened readily for KRAS and V600E mutations to assess their eligibility to be treated with cetuximab or vermurafenib. Applying NGS to screen patients in such a way could increase the number of potential eligible candidates. Lipson et al (2012) used targeted sequencing in colorectal and non-small cell cancer. They identified genomic alterations with a potential clinical therapeutic option in 52.5% (n=40) colorectal cancer and 71% (n=24) non-small cell lung cancer patients. Moreover, they uncovered two new fusion genes, C2orf44-ALK in colorectal cancer and KIF5B-RET in lung adenocarcinoma. The identification of the C2orf44-ALK in colorectal cancer suggests that there may be an unrecognised group of individuals in colorectal cancer that could potentially be challenged with ALK inhibitors. Importantly, the authors report that it is unlikely that this fusion gene would have been detected by current laboratory service techniques such as immunohistochemistry and reverse transcription PCR. Equally important is the issue of whether potentially toxic drugs should be used in a somewhat speculative way based on NGS data.
Next-generation sequencing is likely to have role in evaluating the resistance mechanisms that are developing in the evolving tumour. The clinical response from patients exposed to treatment is often be overcome by resistance to therapy. One example of this is the identification of the T790M mutation in the EGFR gene in lung adenocarcinomas (Yun et al, 2008). Patients with this mutation develop a resistance to EGFR inhibitors. Using NGS one potentially identifies genes that are putative candidates for a resistance mechanism. Identification of a resistance pathway could result in patients being treated with multiple drugs to concurrently block multiple pathways. Escalating this strategy one could potentially use NGS in patients who have exhausted all treatment options to identify putative genes that could serve as druggable targets. However, treating such patients would be controversial and would require a sufficient number of patients in the context of a clinical trial to generate meaningful results.
A potential future application of NGS involves using this technology to develop a sensitive assay to detect early relapse of disease or as a measure of residual disease burden. Primary cancers with/without metastases can be sequenced to identify specific mutations, including rearrangements, which can subsequently be measured in circulating tumour cells or plasma DNA using targeted NGS or other assays. At least some such mutations should be present monoclonally throughout the primary cancer, so as to avoid situations in which the relapsing tumour originates from a subclone without the mutation. Alternatively, if there is concern that some metastases have lost these mutations or that new, critical mutations (such as those that cause chemotherapy resistance) have arisen, genome-wide NGS can be performed serially on DNA in the blood. It is expected that these methods will enable response to treatment and impending relapse to be predicted. McBride et al (2010) provided one of the early examples of using NGS in this way for patients with breast cancer and osteosarcoma. Advances in this field will require stringent validation in the clinical setting and most likely, assessment through clinical trials.
Discussion
Next-generation sequencing is providing researchers with an unprecedented opportunity to uncover the underlying genetic pathways driving cancer. The technology and analytical methods are continually improving in step with one another, although the main issues in the research setting are currently (i) the calling of variants at <50% frequency in the cancer genome and (ii) the sensitive and specific identification of structural and copy number variants. The short-term challenges for NGS in the cancer diagnostic setting are based on the use of focussed sequencing: to reduce costs; to improve technical simplicity and reliability; to enable sequencing of samples with poor-quality or severely limited DNA; and to develop semi-automated, integrated and reliable analysis software that does not require input from an experienced bioinformatician. We envisage that these challenges will be overcome over a period of a few years. Increasingly, focussed cancer NGS will be replaced by larger-scale analyses in the clinical setting. However, it may be some years before these larger-scale analyses are adopted.
In general, for cancer sequencing, it can be argued that error rates higher than those for germline analysis can be tolerated, because, for example, optimal choice of chemotherapy is poorly predicted by conventional methods such as histology. However, difficulties will also be raised, such as the measurement and clinical utility of mutations that are heterogeneous within the tumour. Research studies must be performed to assess the relevance of a priori important mutations that exist within a minor tumour subclone. Moreover, it can be argued that NGS has the major effect of shifting the bottleneck from tumour analysis to the discovery of useful molecular markers. Although NGS will not identify biomarkers and drug targets directly, it will identify mutations that will guide the development of biomarkers and drug targets. In addition, therapeutic dilemmas will occur increasingly often. Suppose that a cancer is analysed by NGS – either genome-wide or for a large gene panel – and an incidental mutation is found in a gene; suppose further that the gene is rarely mutated in that cancer type and has no known role as a driver mutation, yet a targeted agent against that mutation exists, but for which no known role exists in that cancer type. Should the patient receive that agent? Globally, this scenario might occur many times, but there is no easy way of assessing whether the therapy has been useful. We suggest that there is a need for obligatory reporting of targetable, uncommon mutations in specific cancer types and linking to patient data, including response and outcome. As is true for much personalised medicine, while apparently solving many problems, NGS in clinical practice may in fact solve a few problems and provide many opportunities, yet create a need for complex research projects if its full potential is to be fulfilled.
References
Albers CA, Lunter G, MacArthur DG, McVean G, Ouwehand WH, Durbin R (2011) Dindel: accurate indel calls from short-read data. Genome Res 21: 961–973.
Bao S, Jiang R, Kwan W (2011) Evaluation of next generation sequencing software in mapping and assembly. J Hum Genet 56 (6): 406–414.
Birol I, Jackman SD, Nielsen CB, Quian JQ, Varhol R, Stazyk G, Morin RD, Zhao Y, Hirst M, Schein JE, Hormans DE, Connors JM, Gascoyne RD, Marra MA, Jones SJ (2009) De novo transcriptome assembly with ABySS. Bioinformatics 25 (21): 2872–2877.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364: 2507–2516.
Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, Shi X, Fulton RS, Ley TJ, Wilson RK, Ding L, Mardis ER (2009) BreakDancer: an algorithm for high-resloution mapping of genomic structural variation. Nat Methods 6: 677–681.
Chiang DY, Getz G, Jaffe DB, O’Kelly MJ, Zhao X, Carter SL, Russ C, Nusbaum C, Meyerson M, Lander ES (2009) High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods 6: 99–103.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31: 213–219.
Desai AN, Jere A (2012) Next Generation Sequencing: ready for the clinics? Clin Genet 81: 503–510.
Ewing B, Green P (1998b) Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8: 186–194.
Ewing B, Hilier L, Wendl MC, Green P (1998a) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8: 175–185.
Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald N, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal A, Swanton C (2012) Intratumour heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366: 883–892.
Gilbert MTP, Haselkorn T, Bunce M, Sanchez JJ, Lucas SB, Jewell LD, Van Marck E, Worobey M (2007) The isolation of nucleic acids from fixed, paraffin-embedded tissues-which methods are useful when? PLoS One 2 (6): e 537.
Handsaker RE, Korn JM, Nemesh J, McCarroll SA (2011) Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat Genet 43: 269–276.
Horner DS, Pavesi G, Castrignano T, De Meo PD, Liuni S, Sammeth M, Picardi Pesole G E (2010) Bioinformatics approaches for genomics and post genomics applications of next-generation sequencing. Brief Bioinform 11: 181–197.
Ivakhno S, Royce T, Cox AJ, Evers DJ, Cheetham RK, Tavare S (2010) CNAseg-a novel framework for identification of copy number changes in cancer from second-generation sequencing data. Bioinformatics 26: 3051–3058.
Kerick M, Isau M, Timmermann B, Sultmann H, Herwig R, Krobitsch S, Schaefer G, Verdorfer I, Bartsch G, Klocker H, Lehrach H, Schweiger MR (2011) Targeted high throughput sequencing in clinical cancer Settings: formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med Genomics 4: 68.
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK (2012) VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22: 568–576.
Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK, Ding L (2012) SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics 28: 311–317.
Li H, Durbin R (2009a) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760.
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26: 589–595.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009b) The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079.
Li H, Ruan J, Durbin R (2008) Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18: 1851–1858.
Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, Curran JA, Balasubramanian S, Bloom T, Brennan KW, Donahue A, Downing SR, Frampton GM, Garcia L, Juhn F, Mitchell KC, White E, White J, Zwirko Z, Peretz T, Nechustan H, Soussan-Gutman L, Kim J, Sasaki H, Kim HR, Park SI, Ercan D, Sheehan CE, Ross JS, Cronin MT, Janne PA, Stephens PJ (2012) Identification of ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med 18 (3): 382–384.
Lunter G, Goodson M (2011) Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res 21: 936–939.
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, Tang J, Wu G, Zhang H, Shi Y, Liu Y, Chang Yu, Wang B, Lu Y, Han C, Cheung DW, Yiu SM, Peng S, Xiaoqian Z, Liu G, Liao X, Li Y, Yang H, Wang J, Lam TW, Wang J (2012) SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1 (1): 18.
McBride DJ, Orpana AK, Sotiriou C, Joensuu H, Stephens PJ, Mudie LJ, Hamalainen E, Stebbings LA, Anderson LC, Flanagan AM, Durbecq V, Ignatiadis M, Kallioniemi O, Heckman CA, Alitalo K, Edgren H, Futreal PA, Stratton MR, Campbell PJ (2010) Use of cancer-specific genomic rearrangements to quantify disease burden in plasma from patients with solid tumours. Genes Chromosome Cancer 49 (11): 1062–1069.
Metzker ML (2010) Sequencing technologies—the next generation. Nat Rev Genet 11: 31–46.
Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, Chewaskulyong B, Jiang H, Duffield EL, Watkins CL, Armour AA, Fukuoka M (2009) Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 361: 947–957.
Møller P, Hagen AL, Apold J, Maehle L, Clark N, Fiane B, Løvslett K, Hovig E, Vabø A (2007) Genetic epidemiology of BRCA muataions-family history detects less than 50% of the mutation carriers. Eur J Cancer 43 (11): 1713–1717.
Parla JS, Iossifov I, Grabill I, Spector MS, Kramer M, McCombie WR (2011) A comparative analysis of exome capture. Genome Biol 12: 1–17.
Roth A, Ding J, Morin R, Crisan A, Ha G, Giuliany R, Bashashati A, Hirst M, Turashvili G, Oloumi A, Marra MA, Aparicio S, Shah SP (2012) JointSNVMix: a probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics 28: 907–913.
Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M, Leamon JH, Johnson K, Milgrew MJ, Edwards M, Hoon J, Simons JF, Marran D, Myers JW, Davidson JF, Branting A, Nobile JR, Puc BP, Light D, Clark TA, Huber M, Branciforte JT, Stoner IB, Cawley SE, Lyons M, Fu Y, Homer N, Sedova M, Miao X, Reed B, Sabina J, Feierstein E, Schorn M, Alanjary M, Dimalanta E, Dressman D, Kasinskas R, Sokolsky T, Fidanza JA, Namsaraev E, McKernan KJ, Williams AJ, Roth GT, Bustillo J (2011) An integrated semiconductor device enabling non-optical genome sequencing. Nature 475: 348–352.
Stratton MR, Campbell PJ, Futreal PA (2009) The cancer genome. Nature 458: 719–724.
The Cancer Genome Atlas Research Network (2012a) Comprehensive genomic characterization of squamous cell lung cancers. Nature 489: 519–525.
The Cancer Genome Atlas Research Network (2012b) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330–337.
The Cancer Genome Atlas Research Network (2012c) Comprehensive molecular portraits of human breast tumours. Nature 490: 61–70.
ThorvaldsdÓttir H, Robinson JT, Mesirov JP (2013) Integrative genomics viewer (IGV):high-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192.
Venkatesan BM, Bashir R (2011) Nanopore sensors for nucleic acid analysis. Nat Nanotechnol 6: 615–624.
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nuc Acids Res 38 (16): e164.
Yau C, Mouradov D, Jorissen RN, Colella S, Mirza G, Steers G, Harris A, Ragoussis J, Sieber O, Holmes CC (2010) A statistical approach for detecting genomic aberrations in heterogeneous tumor samples from single nucleotide polymorphism genotyping data. Genome Biol 11: R92.
Ye K, Schulz MH, Long Q, Apweiler R, Ning ZM (2009) Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25: 2865–2871.
Yun CH, Mengwasser KE, Torns AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ (2008) The T790M mutation in EGFR kinase causes drug resistance by increasing the affinty for ATP. Proc Natl Acad Sci USA 105: 2070–2075.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Ulahannan, D., Kovac, M., Mulholland, P. et al. Technical and implementation issues in using next-generation sequencing of cancers in clinical practice. Br J Cancer 109, 827–835 (2013). https://doi.org/10.1038/bjc.2013.416
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.416
Keywords
This article is cited by
-
Short report: Performance evaluation of the Idylla™ KRAS and EGFR mutation tests on paraffin-embedded cytological NSCLC samples
Diagnostic Pathology (2021)
-
Application of Next-Generation Sequencing in Neurodegenerative Diseases: Opportunities and Challenges
NeuroMolecular Medicine (2021)
-
Phase II trial of palbociclib in patients with metastatic urothelial cancer after failure of first-line chemotherapy
British Journal of Cancer (2018)
-
A robust targeted sequencing approach for low input and variable quality DNA from clinical samples
npj Genomic Medicine (2018)
-
Towards precision medicine: discovering novel gynecological cancer biomarkers and pathways using linked data
Journal of Biomedical Semantics (2017)
