
Abstract
Background:
Combined inhibition of platelet-derived growth factor receptor beta signalling and vascular endothelial growth factor promotes vascular normalisation in preclinical models and may lead to increased delivery of chemotherapy to tumour tissue. This phase I/II trial assessed the safety and efficacy of capecitabine plus oxaliplatin (XELOX) plus bevacizumab and imatinib in the first-line treatment of patients with metastatic colorectal cancer.
Methods:
Two dose levels (I/II) were defined: capecitabine 850/1000 mg m−2 twice daily on days 1–14; oxaliplatin 100/130 mg m−2 on day 1; bevacizumab 7.5 mg kg−1 on day 1; imatinib 300 mg day−1 on days 1–21 every 21 days. The primary study endpoint was safety. The phase II secondary endpoint was 6-month progression-free survival (PFS).
Results:
Dose level I was chosen for phase II testing because, even though further dose escalation was permitted by the protocol, gastrointestinal toxicities were considered to be clinically significant. A total of 49 patients were evaluated. The 6-month PFS rate was 76%, median PFS was 10.6 months and median overall survival was 23.2 months. Haematological toxicities were generally mild. Sensory neuropathy and diarrhoea were the most common grade 3 toxicities.
Conclusion:
The combination of XELOX with bevacizumab and imatinib is tolerable and has promising efficacy.
Similar content being viewed by others


Main
Chemotherapy with a fluoropyrimidine plus oxaliplatin or irinotecan combined with agents targeting the epidermal growth factor receptor in K-ras wild-type patients or vascular endothelial growth factor (VEGF) are regarded as standard of care in the treatment of patients with metastatic colorectal cancer (mCRC) (Van Cutsem et al, 2010). Recently, the potential benefits of continuing bevacizumab after disease progression (Grothey et al, 2008) have been confirmed by two phase III trials that showed prolonged progression-free survival (PFS) and/or overall survival (OS) with bevacizumab plus second-line chemotherapy vs chemotherapy alone (Masi et al, 2012; Bennouna et al, 2013). However, the clinical effects were smaller than expected from registration data. Additionally, the antibody construct aflibercept, which targets both VEGF and placental growth factor, improved PFS and OS in combination with chemotherapy vs chemotherapy alone when given as second-line therapy; a subgroup of patients who had received first-line bevacizumab showed a similar benefit (Van Cutsem et al, 2012). Based on these data, it is clear that anti-angiogenic therapy has an important role in mCRC but further improvements are needed.
VEGF receptor (VEGFR) signalling induces neoangiogenesis with a structurally chaotic vasculature characterised by increased leakiness and interstitial fluid pressure (IFP), which impair the delivery of cytotoxic agents to the tumour. Normalisation of the tumour vasculature is considered to be a key mechanism of action of VEGF-targeted therapies (Jain, 2005). Platelet-derived growth factor receptor beta (PDGFRβ) is expressed on different cell types in the tumour stroma (i.e. endothelial cells and pericytes). Accordingly, the PDGFRβ pathway is essential for recruiting perivascular cells during angiogenesis (Pietras et al, 2003). Preclinical models show that immature tumour vessels are more prone to the anti-angiogenic effects of VEGF-targeted treatment (Bergers et al, 2003) and that inhibition of PDGFRβ signalling decreases IFP (Pietras et al, 2002). Additionally, synergistic effects from the combined inhibition of VEGF and PDGFRβ signalling might be expected according to preclinical data (Klosowska-Wardega et al, 2009).
In principle, tyrosine kinase inhibitors (TKI) targeting both VEGFR and PDGFRβ (i.e., sunitinib) are available. However, TKI targeting VEGFR did not show clinical efficacy in mCRC (Sobrero and Bruzzi, 2011; Van Cutsem et al, 2011; Schmoll et al, 2012), and a phase III trial testing sunitinib plus 5-fluorouracil/leucovorin plus irinotecan (FOLFIRI) in mCRC was stopped early due to lack of clinical efficacy (Carrato et al, 2013).
Therefore, we sought to establish a combination therapy of bevacizumab with the TKI imatinib, a powerful inhibitor of PDGFRβ, and capecitabine plus oxaliplatin (XELOX) in a phase I/II trial. The clinical activity of imatinib in blocking PDGFRβ-signalling in perivascular cells has been demonstrated in patients with pulmonary arterial hypertension at doses of 200–400 mg day−1 (Ghofrani et al, 2005; ten Freyhaus et al, 2012) and a dose of 300 mg day−1 is supported by recent pharmacokinetic data (Michael et al, 2013). To the best of our knowledge, these are the first data on the tolerability/toxicity and clinical efficacy of this particular combination in patients with previously untreated mCRC.
Patients and methods
Study design
Study AIO KRK 0205 was a prospective, non-randomized, open-label phase I/II clinical trial conducted in five centres in Germany within the Arbeitsgemeinschaft Internistische Onkologie (AIO) according to the Helsinki Declaration. The protocol was reviewed and approved by the local ethics committees of all participating centres; all patients gave written informed consent and the data were monitored by an independent data safety monitoring board (DSMB). The investigators designed, conducted and analysed the study independently. The trial was registered on ClinicalTrials.gov (identifier: NCT00784446).
The primary objective of phase I was to assess the tolerability, safety and dose-limiting toxicities (DLT) of XELOX plus bevacizumab and imatinib, and to define the recommended doses for phase II study. The phase II primary endpoint was safety and toxicity. Secondary endpoints were: Progression free rate at 6 months; objective response rate; PFS; OS, and rate of secondary resection of metastasis.
A cross-trial comparison of cycle numbers with study AIO KRK 0604 (Schmiegel et al, 2013), which included XELOX plus bevacizumab as one study arm (n=127), was also performed.
Patients
Eligible patients were aged ⩾18 years, had previously untreated, histologically proven unresectable stage IV colorectal cancer. Patients were required to have an Eastern Cooperative Oncology Group (ECOG) performance status <2, and adequate renal, hepatic and haematological function. K-ras mutation status was determined as part of routine clinical practice and was available in 36 of 49 patients (73%).
Chemotherapy
All patients received combination therapy consisting of XELOX plus bevacizumab and imatinib. Capecitabine was administered orally twice daily (bid) on days 1 (evening) to 15 (morning). Oxaliplatin was given intravenously as a 2-h infusion on day 1. Bevacizumab was given intravenously as a 30–90-min infusion on day 1. Imatinib was administered orally once daily on days 1–21. The treatment cycle duration was 21 days. Treatment was administered until disease progression or intolerable toxicity.
The following dose levels were defined for phase I dose escalation: dose level I, capecitabine 850 mg m−2 bid, oxaliplatin 100 mg m−2, bevacizumab 7.5 mg kg−1 and imatinib 300 mg day−1; dose level II, capecitabine 1000 mg m−2 bid, oxaliplatin 130 mg m−2, bevacizumab 7.5 mg kg−1 and imatinib 300 mg day−1.
Dose modifications were performed according to the study protocol to manage adverse events. For toxicities that led to the termination of one or two agents, only the following combinations were allowed: capecitabine+oxaliplatin±bevacizumab; or capecitabine+bevacizumab±imatinib. Monotherapy was not permitted.
Assessments
Toxicities were assessed using the Common Terminology Criteria for Adverse Events (CTCAE), version 3.0. Dose-limiting toxicity (DLT) was defined as grade 3 or 4 leukopenia or neutropenia with complications (i.e. fever); grade 4 neutropenia or leukopenia or grade 3 or 4 thrombocytopenia lasting >7 days; grade 3 gastrointestinal toxicity (i.e. diarrhoea, mucositis) lasting >4 days despite adequate supportive care; all grade 4 gastrointestinal toxicities; grade 3 or 4 (in case of liver metastasis) hepatic toxicity; grade 3 or 4 acute or chronic sensory neuropathy; grade 3 hand-foot-skin reactions; other organ toxicities of >2 in severity, with the exception of medically irrelevant events (i.e. alopecia etc.) and allergic reactions to bevacizumab. All toxicities resulting in discontinuation of study medication were also defined as DLT.
For the assessment of tumours, computed tomography scans were performed at baseline, 6 weeks after the first cycle and every 9 weeks thereafter during treatment. Responses were analysed by the investigators according to Response Evaluation Criteria In Solid Tumours (RECIST), version 1.0. Progression-free survival was defined as the interval between start of treatment and the occurrence of progression, diagnosis of a second cancer, or death from any cause. Overall survival was defined as the interval between start of treatment and death from any cause.
Statistical considerations
Dose escalation followed a 6+3 design, that is 6 patients were initially treated at dose level I. If 0–1 patients experienced DLT from this cohort, the next dose level was used. If 2–3 patients experienced DLT at dose level I, a further 3 patients were treated at this dose level. If 2–3 of 9 patients experienced DLTs at dose level I, dose level II was initiated.
In phase II, a one-step design according to Fleming (1982) was used and sample size calculation was based on the assumption that a 6-month PFS ⩾75% was promising contrasting to a futile rate of 60%. Based on a type I error of α=10% and power of 80%, 44 patients had to be included in the study.
Analysis of all parameters was performed descriptively, giving frequencies, means, medians, ranges and confidence intervals (CI). Median PFS and OS were estimated using Kaplan–Meier methods and expressed with 95% CI.
For the cross-trial comparison of the number of cycles with AIO KRK 0604, a two-tailed Wilcoxon–Mann–Whitney U-test was used and Hodges–Lehmann estimator was applied to calculate the probability of a difference between trials.
Results
Fifty-one patients were enrolled between April 2008 and February 2010. Two patients were considered ineligible because of major protocol violations (discontinuation of chemotherapy after one cycle and withdrawal of informed consent, n=1; pre-existing coronary heart disease, n=1). Therefore, 49 patients were evaluable according to intention-to-treat (ITT) principles. Patient characteristics (n=49) are given in Table 1.
Treatment
A total of 585 cycles of study treatment were administered (median 10, range 1–36). The median duration of treatment was 6.2 months (range 0.4–24.4). Dose reductions/discontinuation of drugs according to the combinations prespecified in the study protocol (see Patients and Methods) were required by 30 (61%) patients, most commonly because of adverse events. Adjustments were required for oxaliplatin, capecitabine, imatinib and bevacizumab in 52 (9%), 53 (9%), 43 (7%) and 21 (4%) cycles, respectively. A simplified combination of capecitabine plus bevacizumab plus imatinib was applied in seven patients for a total of 44 cycles (median 3, range 1–17).
The cross-trial comparison revealed that the median number of cycles in the AIO KRK 0604 trial with XELOX plus bevacizumab was 8 vs 10 cycles in the present trial. In AIO KRK 0604, 15% of patients received ⩾14 cycles and 9% received ⩾20 cycles. In the present trial, 31% of patients received ⩾14 cycles and 16% of patients received ⩾20 cycles (Hodges–Lehmann estimator 0.575, 95% CI: 0.483–0.667, P=0.12).
Toxicity
Dose level I was expanded to nine patients after two of the six initial patients developed grade 3 toxicity (diarrhoea/vomiting, n=1; paraesthesia n=1). One patient in the next cohort of three patients also developed grade 3 diarrhoea and vomiting. Thus at dose level I, three of nine patients experienced DLT. Despite the fact that further dose escalation was permitted according to the study protocol, the main toxicities at dose level I (diarrhoea and vomiting) were considered to be clinically significant by the DSMB. Dose level I was therefore chosen for phase II study for safety reasons despite the fact that the maximum-tolerated dose (MTD) was not formally reached.
In the total phase I/II study population, 49 patients were evaluable for toxicity. A summary of toxicities is provided in Table 2. Haematological toxicity was moderate with grade 3 leukopenia observed in one patient (2%) and grade 3 neutropenia in two patients (4%). There were no grade 4 haematological toxicities; however, febrile neutropenia occurred in one patient. Gastrointestinal toxicities were more common; grade 3 diarrhoea developed in eight patients (16%) and vomiting in three patients (6%). Grade 3 sensory neuropathy occurred in 13 patients (27%); no grade 4 toxicity was observed. The most common bevacizumab-related toxicity was grade 1/2 hypertension which occurred in seven patients (14%). Furthermore, there were three grade 2/3 thromboembolic events, and one patient developed grade 3 vascular leak syndrome. Hand-foot-skin reactions occurred in 21 patients (42%), but only 4% were grade 3 events. There were no treatment-related deaths.
Efficacy
Forty-nine patients were evaluable for efficacy. One patient had a complete response, 21 patients had partial responses, 20 patients had stable disease and 3 patients had progressive disease. In four patients, the response was unknown because of withdrawal of consent or other reasons (i.e. toxicity). Thus, the overall response rate (complete plus partial response) was 45% (22 of 49 patients), and the disease control rate (complete plus partial response plus stable disease) was 86% (42 of 49 patients) (Table 3). Potentially curative resections of metastases were performed in 4 out of 48 (8%) patients.
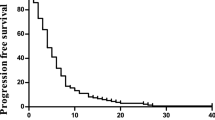
In the ITT population (n=49), 37 (76%) patients were progression-free at 6 months. Median PFS (ITT population) was 10.6 months (95% CI 9.5–15.8 months; Figure 1). Median OS (ITT population) was 23.2 months (95% CI 18.8–31.1 months; Figure 1). Survival endpoints according to K-ras mutation status revealed a median PFS of 9.7 months in patients with K-ras wild-type tumours (n=22) and 10.4 months in patients with K-ras mutated tumours (n=14), and median OS of 26.7 and 24.1 months, respectively.

Probability of PFS and OS.
Discussion
In this phase I/II trial, the safety and tolerability of a combination of XELOX with bevacizumab and imatinib was studied. This combination was chosen to provide improved anti-angiogenic activity by blocking both the VEGF and PDGFRβ signalling pathways. A standard dose of bevacizumab was used, and a dose of imatinib 300 mg day−1 was applied based on the results of pharmacokinetic studies in patients with chronic myeloic leukaemia indicating that doses ⩾300 mg daily are sufficient to inhibit PDGFRβ signalling in vivo (Peng et al. 2004). Furthermore, clinical data from patients with pulmonary hypertension have shown that doses of 200–400 mg day−1 are sufficient to achieve PDGFRβ inhibition in vivo (Ghofrani et al, 2005; ten Freyhaus et al, 2012). A formal pharmacokinetic assessment was beyond the scope of the current trial. However, a recent pharmacokinetic study of 5-fluorouracil/leucovorin plus oxaliplatin (FOLFOX-6) with bevacizumab plus imatinib supports the imatinib dosing schedule (300 mg day−1) (Michael et al, 2013). Further, dose reductions to 200 mg day−1 were required in only 7% of cycles in the present trial.
In the phase I part of the study, gastrointestinal toxicity was the main DLT (i.e. diarrhoea and vomiting) and dose level I was chosen for phase II because these toxicities were considered to be clinically relevant, despite the fact that the criteria for MTD had not formally been met. Dose level I included a 15% decrease in the dose of capecitabine and a 23% decrease in the dose of oxaliplatin compared to standard XELOX, while bevacizumab was used at a standard dose (7.5 mg kg−1 every 3 weeks). Haematological toxicity was infrequent. This contrasts with another phase I trial where imatinib doses of 400 and 600 mg day−1 were used together with FOLFOX-6 (Michael et al, 2013). Thus, the dose of imatinib 300 mg day−1 appears to be a good compromise between PDGF-inhibiting activity and toxicity. Gastrointestinal toxicity was more common. At the dose level used the incidence and grade of diarrhoea, vomiting and nausea was within the expected range compared to other phase II and III trials using XELOX plus bevacizumab (Saltz et al, 2008; Tol et al, 2009; Schmiegel et al, 2013). Grade 2/3 HFS occured in 11 (22.4%) patients compared to a rate of 16% grade 3/4 HFS (Schmiegel et al, 2013) and 19.4% of grade 3/4 HFS (Tol et al, 2009) reported in other trial. Despite the reduced dose of oxaliplatin used, the frequency of grade 3 peripheral neuropathy (27%) was higher than in other clinical trials using oxaliplatin combinations plus bevacizumab. Another AIO trial recently reported a rate of 24% grade 3/4 polyneuropathy (Schmiegel et al, 2013) and in phase III trials rates of 18% (Saltz et al, 2008) and 10.4% (Tol et al, 2009) were reported, respectively. It remains unclear if combined VEGF and PDGFRβ inhibition might be responsible for this effect by interacting with nerve function during oxaliplatin-induced neurotoxicity. This toxicity limits prolonged use of this combination during first-line treatment. Side effects related to bevacizumab (i.e. hypertension and thromboembolic events) were in the expected range, and the addition of imatinib did not appear to aggravate these events. The same was true for other side effects like fatigue.
Efficacy is promising with a 6-month PFS rate of 76%, a median PFS of 10.6 months and median OS of 23.2 months. These results compare favourably with the findings of a phase III trial (median PFS 9.4 months, median OS 21.4 months) that tested XELOX or FOLFOX plus bevacizumab (Saltz et al, 2008), and are similar to another phase III trial (10.7 and 20.3 months, respectively) (Tol et al, 2009) and the German phase II trial AIO KRK 0604 (10.4 and 26.7 months, respectively) (Schmiegel et al, 2013) that both tested XELOX plus bevacizumab. It is important, however, to take into account that the XELOX doses were lower in the present trial. Furthermore, in a cross-trial comparison between AIO KRK 0604 (Schmiegel et al, 2013) and this trial, a statistical trend towards the application of more treatment cycles in the present trial was noted. This may also be related to the fact that the protocol of this trial allowed prespecified dose reductions and discontinuation of drugs (i.e. oxaliplatin) due to toxicity, and patients could benefit from continuation of a less-toxic regimen with capecitabine plus bevacizumab/imatinib. In line with this, a phase I dose-escalation study using 5-fluorouracil plus imatinib in gastric cancer patients reported a favourable toxicity profile (Al-Batran et al, 2007).
It is of interest that results from trials using TKI for the inhibition of VEGF or combined inhibition of VEGF and PDGFRβ in mCRC were disappointing. In a Japanese phase II trial of FOLFIRI plus sunitinib in patients with mCRC, the median PFS was short (6.7 months) (Tsuji et al, 2012), and a phase III trial of FOLFIRI with or without sunitinib was stopped early due to lack of efficacy (Carrato et al, 2013). Besides differences in the toxicity profiles, an optimal ratio in blocking different angiogenic pathways may be required to achieve good clinical results. This may not have been the case for some TKI (i.e. sunitinib).
Conclusion
XELOX plus bevacizumab and imatinib at the doses defined is a clinically feasible regimen. Oxaliplatin-related neurotoxicity may be slightly aggravated by combined VEGF and PDGFRβ inhibition. Efficacy data are promising, especially in view of the reduced chemotherapy doses. Combined inhibition of VEGF and PDGFRβ using bevacizumab plus imatinib is a concept worth investigating in further clinical trials, especially as maintenance therapy in combination with a fluoropyrimidine alone.
Change history
17 September 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Al-Batran SE, Atmaca A, Schleyer E, Pauligk C, Hosius C, Ehninger G, Jäger E (2007) Imatinib mesylate for targeting the platelet-derived growth factor beta receptor in combination with fluorouracil and leucovorin in patients with refractory pancreatic, bile duct, colorectal, or gastric cancer-a dose-escalation phase I trial. Cancer 109: 1897–1904.
Bennouna J, Sastre J, Arnold D, Österlund P, Greil R, Van Cutsem E, von Moos R, Viéitez JM, Bouché O, Borg C, Steffens CC, Alonso-Orduña V, Schlichting C, Reyes-Rivera I, Bendahmane B, André T, Kubicka S ML18147 Study Investigators (2013) Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol 14: 29–37.
Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D (2003) Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest (2003) 111: 1287–1295.
Carrato A, Swieboda-Sadlej A, Staszewska-Skurczynska M, Lim R, Roman L, Shparyk Y, Bondarenko I, Jonker DJ, Sun Y, De la Cruz JA, Williams JA, Korytowsky B, Christensen JG, Lin X, Tursi JM, Lechuga MJ, Van Cutsem E (2013) Fluorouracil, leucovorin, and irinotecan plus either sunitinib or placebo in metastatic colorectal cancer: a randomized, phase III trial. J Clin Oncol 31: 1341–1347.
Fleming TR (1982) One-sample multiple testing procedure for phase II clinical trials. Biometrics 38 (1): 143–151.
Ghofrani HA, Seeger W, Grimminger F (2005) Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med 353: 1412–1413.
Grothey A, Sugrue MM, Purdie DM, Dong W, Sargent D, Hedrick E, Kozloff M (2008) Bevacizumab beyond first progression is associated with prolonged overall survival in metastatic colorectal cancer: results from a large observational cohort study (BRiTE). J Clin Oncol 26: 5326–5334.
Jain RK (2005) Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307: 58–62.
Klosowska-Wardega A, Hasumi Y, Burmakin M, Ahgren A, Stuhr L, Moen I, Reed RK, Rubin K, Hellberg C, Heldin CH (2009) Combined anti-angiogenic therapy targeting PDGF and VEGF receptors lowers the interstitial fluid pressure in a murine experimental carcinoma. PLoS One 4: e8149.
Köhne CH, Cunningham D, Di Costanzo F, Glimelius B, Blijham G, Aranda E, Scheithauer W, Rougier P, Palmer M, Wils J, Baron B, Pignatti F, Schöffski P, Micheel S, Hecker H (2002) Clinical determinants of survival in patients with 5-fluorouracilbased treatment for metastatic colorectal cancer: results of a multivariate analysis of 3825 patients. Ann Oncol 13: 308–317.
Masi G, Loupakis F, Salvatore L, Cremolini C, Fornaro L, Schirripa M, Fea E, Granetto C, Antonuzzo L, Giommoni E, Allegrini G, Cupini S, Boni C, Banzi M, Chiara S, Sonaglio C, Valsuani C, Bonetti A, Boni L, Falcone A (2012) A randomized study evaluating the continuation of bevacizumab beyond progression in metastatic colorectal cancer patients who received bevacizumab as part of first-line treatment: results of the BEBYP trial by the Gruppo Oncologico Nord Ovest (GONO). Ann Oncol 23 (suppl 9) ixe9: LBA17.
Michael M, Zalcberg J, Gibbs P, Lipton L, Gouillou M, Jefford M, McArthur G, Copeman M, Lynch K, Tebbutt NC (2013) A phase I trial of imatinib in combination with mFOLFOX6-bevacizumab in patients with advanced colorectal cancer. Cancer Chemother Pharmacol 71: 321–330.
Peng B, Hayes M, Resta D, Racine-Poon A, Druker BJ, Talpaz M, Sawyers CL, Rosamilia M, Ford J, Lloyd P, Capdeville R (2004) Pharmacokinetics and pharmacodynamics of imatinib in a phase I trial with chronic myeloid leukemia patients. J Clin Oncol 22: 935–942.
Pietras K, Sjoblom T, Rubin K, Heldin CH, Ostman A (2003) PDGF receptors as cancer drug targets. Cancer Cell 3: 439–443.
Pietras K, Rubin K, Sjoblom T, Buchdunger E, Sjöquist M, Heldin CH, Ostman A (2002) Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res 62: 5476–5484.
Saltz LB, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, Koski S, Lichinitser M, Yang TS, Rivera F, Couture F, Sirzén F, Cassidy J (2008) Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 26: 2013–2019.
Schmiegel W, Reinacher-Schick A, Arnold D, Kubicka S, Freier W, Dietrich G, Geissler M, Hegewisch-Becker S, Tannapfel A, Pohl M, Hinke A, Schmoll HJ, Graeven U (2013) Capecitabine/irinotecan or capecitabine/oxaliplatin in combination with bevacizumab is effective and safe as first-line therapy for metastatic colorectal cancer: a randomized phase II study of the AIO colorectal study group. Ann Oncol 24 (6): 1580–1587.
Schmoll HJ, Cunningham D, Sobrero A, Karapetis CS, Rougier P, Koski SL, Kocakova I, Bondarenko I, Bodoky G, Mainwaring P, Salazar R, Barker P, Mookerjee B, Robertson J, Van Cutsem E (2012) Cediranib with mFOLFOX6 versus bevacizumab with mFOLFOX6 as first-line treatment for patients with advanced colorectal cancer: a double-blind, randomized phase III study (HORIZON III). J Clin Oncol 30: 3588–3595.
Sobrero AF, Bruzzi P (2011) Vatalanib in advanced colorectal cancer: two studies with identical results. J Clin Oncol 29: 1938–1940.
ten Freyhaus H, Dumitrescu D, Berghausen E, Vantler M, Caglayan E, Rosenkranz S (2012) Imatinib mesylate for the treatment of pulmonary arterial hypertension. Expert Opin Investig Drugs 21: 119–134.
Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, Erdkamp FL, Vos AH, van Groeningen CJ, Sinnige HA, Richel DJ, Voest EE, Dijkstra JR, Vink-Börger ME, Antonini NF, Mol L, van Krieken JH, Dalesio O, Punt CJ (2009) Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med 360: 563–572.
Tsuji Y, Satoh T, Tsuji A, Muro K, Yoshida M, Nishina T, Nagase M, Komatsu Y, Kato T, Miyata Y, Mizutani N, Hashigaki S, Lechuga MJ, Denda T (2012) First-line sunitinib plus FOLFIRI in Japanese patients with unresectable/metastatic colorectal cancer: a phase II study. Cancer Sci 103: 1502–1507.
Van Cutsem E, Bajetta E, Valle J, Köhne CH, Hecht JR, Moore M, Germond C, Berg W, Chen BL, Jalava T, Lebwohl D, Meinhardt G, Laurent D, Lin E (2011) Randomized, placebo-controlled, phase III study of oxaliplatin, fluorouracil, and leucovorin with or without PTK787/ZK 222584 in patients with previously treated metastatic colorectal adenocarcinoma. J Clin Oncol 29: 2004–2010.
Van Cutsem E, Nordlinger B, Cervantes A ESMO Guidelines Working Group (2010) Advanced colorectal cancer: ESMO Clinical Practice Guidelines for treatment. Ann Oncol 21 (Suppl 5): v93–v97.
Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausová J, Macarulla T, Ruff P, van Hazel GA, Moiseyenko V, Ferry D, McKendrick J, Polikoff J, Tellier A, Castan R, Allegra C (2012) Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol 30: 3499–3506.
Acknowledgements
The authors thank Dr A. Kranich and her team (GSO Hamburg, Germany) for their logistical support during the study. Furthermore, we would like to thank Lars Pester and Anna Kostenko from the study team in Cologne for their important contribution to the conduct of this clinical trial. We thank Florian Lordick for helpful advice after carefully reading the manuscript. This work was supported by Roche Pharma AG, Novartis and Sanofi-Aventis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
AH has received research support from Novartis; DA, RH and UTH have received research sponsorship from Roche; DA has received speaker’s honoraria from Roche and Sanofi-Aventis; RH and UTH have received speaker’s honoraria from Roche. The remaining authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Hoehler, T., von Wichert, G., Schimanski, C. et al. Phase I/II trial of capecitabine and oxaliplatin in combination with bevacizumab and imatinib in patients with metastatic colorectal cancer: AIO KRK 0205. Br J Cancer 109, 1408–1413 (2013). https://doi.org/10.1038/bjc.2013.409
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.409
