
Abstract
Objective:
We have previously identified peroxiredoxin-3 (PRDX-3) as a cell-surface protein that is androgen regulated in the LNCaP prostate cancer (PCa) cell line. PRDX-3 is a member of the peroxiredoxin family that are responsible for neutralising reactive oxygen species.
Experimental design:
PRDX-3 expression was examined in tissue from 32 patients using immunohistochemistry. Subcellular distribution was determined using confocal microscopy. PRDX-3 expression was determined in antiandrogen-resistant cell lines by western blotting and quantitative RT–PCR. The pathways of PRDX-3 overexpression and knockdown on apoptosis and response to oxidative stress were investigated using protein arrays.
Results:
PRDX-3 is upregulated in a number of endocrine-regulated tumours; in particular in PCa and prostatic intraepithelial neoplasia. Although the majority of PRDX-3 is localised to the mitochondria, we have confirmed that PRDX-3 at the cell membrane is androgen regulated. In antiandrogen-resistant LNCaP cell lines, PRDX-3 is upregulated at the protein but not RNA level. Resistant cells also possess an upregulation of the tricarboxylic acid (TCA) pathway and resistance to H2O2-induced apoptosis through a failure to activate pro-apoptotic pathways. Knockdown of PRDX-3 restored H2O2 sensitivity.
Conclusion:
Our results suggest that PRDX-3 has an essential role in regulating oxidation-induced apoptosis in antiandrogen-resistant cells. PRDX-3 may have potential as a therapeutic target in castrate-independent PCa.
Similar content being viewed by others


Main
The mammalian peroxiredoxins (PRDXs) are a highly conserved family of thiol-containing peroxidises that has six members (PRDX1–6) (Leyens et al, 2003; Hall et al, 2009). The PRDX proteins catalyse the reduction of molecules that cause oxidative stress such as reactive oxygen species (ROS), for example, peroxides that are essential metabolic intermediates and regulators of growth factor signalling but are often produced as a result of cellular stress (Wood et al, 2003; Giorgio et al, 2007; Cox et al, 2009; Lenaz, 2012; Miki and Funato, 2012; Ray et al, 2012). If left unchecked, ROS can damage DNA and induce tumourigenesis (Trachootham et al, 2008; Ruckenstuhl et al, 2009). Alternatively, the induction of ROS formation by chemotherapy and ionising radiation can be used therapeutically to cause DNA damage-induced cell death (Lee et al, 2011). Catalysis of ROS leads to oxidised SO2 and SO3 forms of the PRDX proteins which can be reduced, and therefore reactivated, via a thioredoxin and ATP-dependent mechanism (Fujii and Ikeda, 2002; Aran et al, 2009; Hanschmann et al, 2010). The PRDX protein family have previously been linked to cancer development (Shen and Nathan, 2002). All of the PRDX proteins that have been linked to the development or progression of tumourigenesis as summarised in Table 1. PRDX-1, 2, 5 and 6 have also been linked to resistance to chemotherapy, in particular cisplatin, and ionising radiation, in a variety of tumours including ovarian, oesophageal and hepatocellular (Kikuta et al, 2010; Dittmann et al, 2011; Gao et al, 2011; Lee et al, 2011; Pak et al, 2011; Cho et al, 2012; Zhiyu et al, 2012). Increased PRDX-2 is thought to induce resistance by preventing DNA damage-induced apoptosis (Lee et al, 2011).
Prostate cancer (PCa) is the most prevalent male cancer in the Western world and it requires androgens such as testosterone or dihydrotestosterone (DHT) for continued growth and development (Friedlander and Ryan, 2012). Treatment for PCa targets this reliance on androgens by blocking androgen production or the androgen receptor (Kuil and Mulder, 1994). However, prostate cells eventually escape this androgen blockade and continue to grow, becoming resistant to therapy, despite continued antiandrogen treatment. The mechanisms involved in the progression to castrate-independent disease remains poorly understood, but may include androgen receptor mutation or amplification, receptor activation by alternative ligands such as oestrogen, changes in cofactor expression or alterations in growth factor signalling (Bevan, 2005). Second line chemotherapy with docetaxel is only effective in 30–50% of patients (Tannock et al, 2004). PRDX-1 is known to interact with the androgen receptor, enhancing ligand binding and regulating transactivation of androgen-regulated genes (Park et al, 2007; Chhipa et al, 2009; Shiota et al, 2011; Miki and Funato, 2012; Ray et al, 2012).
PRDX-3 is a known c-myc, miR-383 and miR-23b target gene required for mitochondrial homoeostasis and neoplastic transformation (Wonsey et al, 2002; Vivas-Mejia et al, 2009; He et al, 2012; Li et al, 2012). A high level of expression is found in the breast, cervical, hepatocellular and prostate carcinomas (Noh et al, 2001; Choi et al, 2002; Wonsey et al, 2002; Karihtala et al, 2003; Lin et al, 2007; Kim et al, 2009; Basu et al, 2011). In PCa, the expression of PRDX-3 has been negatively correlated with TMPRSS–ERG fusion status and overexpression has been shown to increase cellular proliferation regardless of the hormone dependency of the cell line (Ummanni et al, 2012). The subcellular localisation of the different PRDX proteins is diverse (Fujii and Ikeda, 2002). The majority of PRDX-3 has been localised to the mitochondria with a small proportion also reported at the cell surface, similar to reports of membrane-associated PRDX-6 (Araki et al, 1999; Liu et al, 2005; Whitaker et al, 2007; Ambruso et al, 2012). PRDX-3 expression is induced by oxidants in the cardiovascular system (Araki et al, 1999; Kumar et al, 2009) and is particularly sensitive to oxidative stress following treatment with pro-apoptotic drugs, such as auranofin, isothiocyanates and arsenic trioxide (Brown et al, 2008; Cox et al, 2008a; Vivas-Mejia et al, 2009).
Using immunohistochemistry (IHC), we have shown upregulation of PRDX-3 in endocrine-regulated tumours and in particular prostatic intraepithelial neoplasia (PIN) and PCa. PRDX-3 protein, but not mRNA, is significantly upregulated in antiandrogen-resistant PCa cell lines, resulting in increased resistance to oxidative stress and failure to activate pro-apoptotic pathways. Conversely, knockdown of PRDX-3 leads to raised susceptibility to oxidative stress. These results suggest that PRDX-3 may be essential for the development of PCa and the development of resistance to treatment.
Materials and methods
Patient cohorts
Prostate tissue from radical prostatectomies performed at Addenbrookes Hospital, Cambridge, UK between 2001 and 2005 was used to make tissue microarrays (TMAs) using duplicate 0.6-mm cores taken from paraffin-embedded tissue and a Beecher Manual TMA Arrayer. Details of this array have previously been published (Whitaker et al, 2010). In total, the tissue from 32 different patients was used to generate the TMA. Regions of benign or normal prostate (n=4), PIN (n=4) and malignancy (n=2–6) were identified by a specialist uro-pathologist (AYW) for each patient (Whitaker et al, 2010). Malignant tissue was obtained from at least one and, where possible, up to three different tumour foci from each patient. Stage and Gleason grade was confirmed by a specialist uro-pathologist (AYW) before scoring any IHC staining. Full ethical approval was obtained before beginning this study.
IHC and image analysis
A TMA containing one matched normal and one tumour core from a variety of different organs (Stretton Scientific, Stretton, UK) or the prostate TMA outlined above was probed with anti-PRDX-3 antibody (1 : 1000, Abcam, Cambridge, UK) using a Bondmax Autostainer. PRDX-3 staining was visualised using Alexafluor 488 (green) (Molecular Probes, Paisley, UK) and counterstained with DAPI (blue) to visualise nuclei. The PRDX-3-stained prostate TMAs were scanned and quantified using the Ariol system (Applied Imaging, New Milton, UK). Cores containing mixed pathology or only stroma were excluded from the analysis. A threshold intensity of 75 was applied to all images to remove pixel densities caused by non-specific staining. After cores were mapped, total pixel count for each core was calculated using the following equation: total pixel intensity × total pixel area/1000. Data were grouped according to pathology, for example, benign, PIN or PCa or subdivided by Gleason grade. N numbers indicate the number of cores analysed. P-values were calculated using a Kruskal–Wallis test.
Cell culture
COS Cells derived from Monkey kidney and human prostate PC3 and parental LNCaP cells were purchased from the Cancer Research UK cell bank. Primary benign prostatic hyperplasia fibroblasts were a kind gift from Dr Michael Brown (The Genito-Urinary Cancer Research Group, Cancer Research UK Paterson Institute for Cancer Research). COS cells were routinely cultured in DMEM supplemented with 10% foetal bovine serum (Labtech, Uckfield, UK). All other cells lines were routinely cultured in RPMI media (Gibco, Paisley, UK) with 10% foetal bovine serum. Antiandrogen-resistant LNCaP cells have previously been characterised (Hobisch et al, 2006; Vias et al, 2006) and were grown in media supplemented with either 1 μ M hydroxyflutamide alone (LNCaP-OHF), 1 μ M OHF and 10 pM R1881 (a non-hydrolysable DHT analogue) (LNCaP-OHF/R1881), 1 μ M alone (LNCaP-BIC), 1 μ M BIC and 10 pM R1881 (LNCaP-BIC/R1881). Short-term antiandrogen treatment was performed using parental LNCaP cells grown in the presence of 0 μ M, 1 μ M or 10 μ M bicalutamide. PRDX-3 knockdown was performed on six-well plates of parental LNCaP cells using 0 μ M, 50 μ M or 100 μ M Accell SMARTpool siRNA (Dharmacon, Loughborough, UK) according to the manufacturer’s guidelines. Cells were harvested or used for additional experiments 72 h after transfection.
Western blotting
Cells were washed in PBS, pelleted and protein lysate was made as before (Whitaker et al, 2008). Twenty micrograms of total protein, as determined using Bradford assay, were compared using the SDS–PAGE and western blot analysis. The following antibodies were used for the detection of proteins: mouse anti-PRDX-3 antibody (1 : 3000), rabbit anti-PRDX-SO2/SO3 (1 : 3000) and rabbit anti-COX IV (1 : 5000) (all from Abcam). Mouse anti-actin (1 : 5000, Abcam) was used as a loading control. Proteins were visualised using anti-mouse or anti-rabbit HRP-conjugated secondary antibodies (1 : 1000, Dako Cytomation, Ely, UK) and ECL-Plus (GE Healthcare, Amersham, UK). If the signal detected was beyond the dynamic range of film, diaminobenzidine (Vector Laboratories, Peterborough, UK) was used for the detection.
Synthesis of cDNA and quantitative real-time PCR
For RNA, cells were harvested in 1 ml Trizol. From each condition, 5 μg of total RNA was reverse transcribed using the SuperScript III First-Strand synthesis system (Invitrogen, Paisley, UK) with random hexamer primers according to manufacturer’s recommendations. For RT–PCR, primers were designed to recognise PRDX-3; forward 5′-GCCGCTCTGTGGATGAGACT-3′, reverse 5′-CCAGCTGGGCACACTTCC-3′. Real-time PCR was performed in triplicate in 10-μl reactions containing 5 μl of SYBR Green PCR Master Mix (Applied Biosystems), 2 pmol of primers and 1.5 μl of cDNA as template. The cycling conditions for the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) were 50 °C for 2 min, 95 °C for 10 min, 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Real-time efficiencies were calculated from the slopes in the standard dilution curves. Relative expression levels were calculated on the basis of the difference in Ct values between the test samples and the control using the following formula: 10 mean-Y intercept/slope. Results were normalised with the expression of GAPDH results expressed relative to vehicle-treated cells.
Gene expression experiments
Gene expression data were generated as part of a previously published study (Massie et al, 2011). Briefly, gene expression analysis was carried out on Illumina Human HT12 version 4 arrays. All analyses were carried out on R using Bioconductor packages (Gentleman, 2004). Raw intensity data were processed using the BASH and HULK algorithms as implemented in the beadarray package (Cairns, 2008; Dunning, 2007). Log2 transformation and quantile normalisation of the data were performed across all sample groups. The differential expression analysis was carried out using the limma package (Smyth, 2005). Points were calculated to represent log-expression relative to the 0 time point. The probability of seeing that if there were not a trend was calculated from 1 000 000 permutations to give the P-value for the hypothesis that there is no change after androgen treatment.
Confocal microscopy
Immunofluorescence was performed as previously described (Whitaker et al, 2007). LNCaP cells were probed for PRDX-3 (1 : 400) in 10% whole-goat serum (Zymed, Paisley, UK) in PBS. For colocalisation, experiments COX IV (1 : 400) and E-cadherin (1 : 400, Abcam) were used. Proteins were visualised using AlexaFluor 488 or 594-conjugated secondary antibodies (Molecular Probes) and visualised using a Nikon Eclipse confocal microscope using a × 100 objective. All scale bars represent 10 μ M.
Citrate synthase assay
Parental LNCaP cells and antiandrogen-resistant cell lines were washed in ice-cold PBS and lysed on ice using CellLytic MT Cell Lysis Reagent (Sigma, St Louis, MO, USA). Ten micrograms total protein was assayed immediately for citrate synthase activity using a Citrate Synthase Assay Kit (Sigma) in accordance with the manufacturer’s instructions. Citrate synthase activity was measured by a change in OD at 412 nm every 10 s for a total of 100 s using a Lucy II spectrophotometer. After plotting A412 against time, the linear range of the plot was used to calculate the citrate synthase activity in μmole ml−1 min−1. P-values were determined using a two-way ANOVA test.
Cell viability assays
Parental LNCaP and resistant LNCaP-BIC cells were grown in six-well dishes to 60% confluence, washed twice in PBS and incubated with serum-free RPMI for 24 h before treatment. Duplicate wells were dosed with 0–0.001% H2O2 for 24 h and cells harvested by scraping into the existing media, centrifuged at 3000 r.p.m. and the media removed. Cells were gently but thoroughly resuspended in ice-cold PBS and cell viability was measured using a ViCell automated cell stainer/counter. Duplicate cells dosed with 0.00033% H2O2 for 24 h were harvested and lysed as for western blotting. Human apoptosis proteome profiler antibody arrays (R&D Systems, Abingdon, UK) were performed in accordance with the manufacturer’s instructions using 200 μg total protein. Following siRNA knockdown for 72 h, LNCaP cells were washed twice in PBS and incubated with serum-free RPMI for 24 h before 0.00033% H2O2 treatment for 24 h. Duplicate cells were scraped, resuspended and cell viability measured as before. All results were calculated as relative to the no treatment controls. P-values were determined using a two-tailed Student’s t-test.
Results
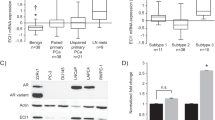
Using a matched tumour and normal TMA containing tissue cores from a variety of organs, we investigated the expression of PRDX-3 using immunofluorescent staining (Figure 1A and Table 2). In this single TMA, PRDX-3 was overexpressed in numerous endocrine tumours such as the thyroid and ovary and highly expressed in both normal and tumourigenic breast. PRDX-3 was also downregulated in many non-endocrine tissues such as the kidney and lung although upregulated in the liver and cervix. Using a prostate TMA, we found a highly significant increase in PRDX-3 in PIN and prostate tumours relative to benign prostate (Figures 1B and C) (P<0.0001). Consistent with previous reports, we found no link between PRDX-3 expression in prostate tumours and Gleason grade (Figure 1D) (P=0.5321) (Basu et al, 2011).

PRDX-3 expression in tissue. A TMA containing matched normal/tumour cores from a variety of different organs was probed with anti-PRDX-3 antibody and visualised using Alexafluor 488 (green). Nuclei were counterstained with DAPI (blue). Examples of different paired normal and tumour tissue are shown (A). A prostate tissue TMA was also stained in an identical manner. Examples from a single patient are shown (B). PRDX-3-stained prostate TMAs were scanned and quantified. Total pixel count for each core was calculated using the following equation: total pixel intensity × total pixel area/1000. Data were grouped according to pathology, for example, benign, prostatic intraepithelial neoplasia (PIN) or prostate cancer (PCa) (C) or subdivided by Gleason grade (D). N numbers indicate the number of cores analysed. P-values were calculated using a Kruskal–Wallis test.
There was no obvious difference in PRDX-3 subcellular distribution between benign and tumourigenic tissue samples (Figure 1B). Tissue staining for PRDX-3 was punctate, consistent with the previously reported mitochondrial localisation (Araki et al, 1999). To confirm that the majority of PRDX-3 was localised to the mitochondria, PRDX-3 was colocalised with COX IV, a known mitochondrial marker, in LNCaP cells (Figure 2A). PRDX-3 has also been localised to the cell surface in a PCa cell line, and we investigated whether it colocalised with E-cadherin, a known plasma membrane protein, using high-power confocal microscopy (Whitaker et al, 2007) (Figure 2B). A proportion of PRDX-3 was seen localised to the cellular membrane alongside E-cadherin. This colocalisation was only visible in vehicle-treated cells and lost in the presence of R1881, confirming the androgen regulation of cell surface PRDX-3 previously reported (Whitaker et al, 2007). Androgen regulation was confirmed using previously published gene expression data, which supported the weak downregulation of PRDX-3 in response to androgen treatment (Figure 2C) (Massie et al, 2011).

PRDX-3 is localised to the cell membrane and can be detected by ELISA. Mitochondrial PRDX-3 was confirmed by colocalisation of COX IV (green) with PRDX-3 (red) (A). For androgen regulation, LNCaP cells were grown in RPMI containing 10% charcoal stripped serum and treated for another 24 h with vehicle or R1881. Cells were fixed with methanol and probed for E-cadherin (green) or PRDX-3 (red). Nuclei are shown in blue. Scale bars represent 10 μ M (B). Gene expression data were generated as previously described in LNCaP cells (Massie, 2011) by treating cells with the androgen, R1881 and harvesting over a timecourse of 24 h. Points represent log-expression relative to the 0 time point. The autocorrelation is 0.61. The probability of seeing that whether there were not a trend is 0.00005 (from 1 000 000 permutations). Hence, this is the P-value for the hypothesis that there is no change after androgen treatment (C).
To investigate how the expression changed following treatment with antiandrogens, PRDX-3 expression was studied in a number of prostate cell lines including the antiandrogen-resistant cells (LNCaP-BIC and LNCaP-OHF) and LNCaP-BIC-R1881 and LNCaP-OHF-R1881 cells cultured with the antiandrogens plus 10 pM of synthetic DHT, R1881 (Hobisch et al, 2006; Vias et al, 2006). Consistent with previous data, total PRDX-3 was downregulated following R1881 treatment of LNCaP cells (Figure 3A) (Whitaker et al, 2007; Massie et al, 2011). Lower expression was also seen in benign fibroblasts and the AR-negative PC3 cells which is consistent with low the PRDX-3 expression in androgen-independent cell lines seen previously (Basu et al, 2011). All of the antiandrogen-resistant cell lines exhibited the upregulation of PRDX-3, compared with vehicle only treated wtLNCaP cells. When PRDX-3 mRNA levels were investigated using qRT–PCR there was a reciprocal correlation with the protein expression with PRDX-3 mRNA increasing with androgen treatment and decreasing in the antiandrogen-resistant cell lines (Figure 3B). To determine whether the changes in PRDX-3 expression had any causal effect in antiandrogen resistance or whether they resulted from antiandrogen resistance, we treated LNCaP cells for 3 days with vehicle, 1 μ M or 10 μ M bicalutamide. Cells treated with bicalutamide quickly became etiolated and showed a more neuroendocrine phenotype consistent with cell stress and the development of resistance (Figure 3D, upper panels) (Vias et al, 2006). PRDX-3 was upregulated in response to bicalutamide in a dose-dependent manner (Figure 3D, lower panel). An antibody that detects the oxidised forms (SO2 and SO3) of PRDX-3 demonstrated a marked upregulation in response to bicalutamide treatment indicating an increase in mitochondrial oxidative stress and PRDX-3 activity.

PRDX-3 is predominantly mitochondrial and upregulated at the protein level by antiandrogen treatment. Twenty micrograms protein lysates from parental LNCaP cells treated with R1881 or vehicle for 24 h, fibroblasts from primary benign prostatic hyperplasia (BPH) COS, PC3 or the antiandrogen-resistant cell lines grown with1 μ M hydroxyflutamide alone (LNCaP-OHF), 1 μ M hydroxyflutamide and 10 pM R1881 (LNCaP-OHF/R1881), 1 μ M bicalutamide alone (LNCaP-BIC), 1 μ M bicalutamide and 10pM R1881 (LNCaP-BIC/R1881) were separated by SDS–PAGE and probed for PRDX-3 and actin (A). qRT–PCR was performed on samples from the same cell lines. Relative expression levels were calculated based on the difference in Ct values between the test samples and the control and results normalised with the expression of GAPDH. All results were expressed relative to vehicle-treated cells. Mean results and s.d. are shown relative to vehicle-treated parental cell lines (*). P-values were calculated using a two-tailed Students t-test (B). Twenty micrograms of identical cell lysates were also analysed by western blotting (WB) for COX IV and actin as before (C). wtLNCaP cells were grown in the presence of 0 μ M, 1 μ M or 10 μ M bicalutamide for 3 days and 20 μg cell lysate separated by SDS–PAGE, transferred to nitrocellulose and probed for oxidized PRDX-3 (SO2/SO3), total PRDX-3 and actin (D, top panel). Images were taken showing the more etiolated appearance of the bicalutamide treated cells (D, lower panels).
To determine whether PRDX-3 is upregulated in antiandrogen-resistant cells or whether there was an increase in overall mitochondrial number or activity, we probed cell lysates for COX IV, a key enzyme in the mitochondrial electron transport chain that is an effective mitochondrial loading control (Figure 2A). COX IV was markedly overexpressed in all of the antiandrogen-resistant cell lines compared with wtLNCaP cells, consistent with the PRDX-3 results and suggestive of increased mitochondrial number. To determine whether this resulted in an increase in the mitochondrial function, we measured the activity of citrate synthase, a key enzyme of the mitochondrial tricarboxylic acid (TCA) cycle (Figure 4A). Citrate synthase activity was increased by R1881 treatment of wtLNCaP cells, whereas the antiandrogen-resistant cells exhibited three- to five-fold greater citrate synthase activity than the androgen-dependent wtLNCaP cells. Thus, long-term antiandrogen-treated cells have more mitochondria and are much more metabolically active than untreated cells (P=0.0032) resulting in an increase in mitochondrial enzymes, including PRDX-3.

PRDX-3 protects cells from apoptosis. For the citrate synthase assay, wtLNCaP cells and antiandrogen-resistant cells were lysed on ice and 10 μg total protein assayed immediately for citrate synthase activity. P-values were determined using a two-way ANOVA test (A). wtLNCaP cells (solid line, squares) and LNCaP-BIC cells (dashed line, circles) were serum starved for 24 h and treated with varying concentrations of H2O2 (0.00016–0.001%) for 24 h before measuring cell viability (B). wtLNCaP cells and LNCaP-BIC (BIC) were serum starved and treated as before with (+) or without (−) 0.00033% H2O2 and 200 μg of total protein used on an apoptosis protein array. Selected changes are highlighted in the lower panel (C). PRDX-3 knockdown was performed on wtLNCaP and LNCaP-BIC cells using 0, 50 or 100 μ M siRNA. Seventy-two hours after transfection cells were harvested, lysed and probed for PRDX-3 or actin (D, right panel). Cells were treated with 0.00033% H2O2 for 1 h or 24 h before harvesting and assaying for cell viability as before (D, left panel). All results were calculated as relative to the no treatment controls. P-values were determined using a two-tailed Student’s t-test.
As PRDX-3 protein increases following antiandrogen treatment, we aimed to establish whether this conferred any survival advantage upon cells that might suggest that PRDX-3 expression might be clinically significant. To induce oxidative stress, both wtLNCaP- and bicalutamide-resistant cells were treated with 0–0.001% H2O2 for 24 h and assayed for cell viability (Figure 4B). Long-term antiandrogen-treated LNCaP-BIC cells with raised PRDX-3 levels showed increased resistance to H2O2-induced oxidative stress, particularly at lower concentrations (0–0.0005%). Cell lysates from wtLNCaP and LNCaP-BIC treated with and without 0.00033% H2O2 were used on an apoptosis focused antibody array to determine the difference in response to oxidative stress (Figure 4C). In these arrays, wtLNCaP and LNCaP-BIC cells with knocked down PRDX-3 (siPRDX3) showed upregulation of a number of pro-apoptotic markers including p21, Fas ligand and cleaved caspase-3, but not Bad. We did not observe any change in the expression of pro-apoptotic proteins in the LNCaP-BIC cells, consistent with the survival advantage seen in Figure 4b. Trail receptor 2 which protects cells against apoptosis was also upregulated in wtLNCaP and LNCaP-BIC siPRDX3 cells but not LNCaP-BIC cells, suggesting deregulation of both pro- and anti-apoptotic pathways. When PRDX-3 was knocked down in wtLNCaP and LNCaP-BIC cells using siRNA, the knocked-down cells were hyper sensitive to treatment with H2O2 compared with a scrambled control (P=0.005 (wtLNCaP), P=0.009 (LNCaP-BIC)) (Figure 4D).
Discussion
Although the PRDX proteins all share peroxidase activity, their expression, particularly in tumours, is diverse (Table 1). Previously PRDX-3 has been shown to be upregulated in a variety of tumour tissues (Noh et al, 2001; Choi et al, 2002; Kinnula et al, 2002; Karihtala et al, 2003; Lehtonen et al, 2004; Lin et al, 2007; Kim et al, 2009; Chua et al, 2010; Basu et al, 2011; Ummanni et al, 2012). Using a matched normal/tumour TMA, we have shown that PRDX-3 upregulation in tumours is most pronounced in endocrine-regulated tissues such as the breast, thyroid and prostate (Figure 1A and Table 2) supporting the previous association made between PRDX-3 upregulation and the presence of hormone receptors in the breast tissue (Karihtala et al, 2003). In PCa, PRDX-3 has been shown to be upregulated in needle-core biopsies and a commercial TMA, where the patient treatment and surgical technique could not be determined (Lin et al, 2007; Basu et al, 2011). Our data, gathered from 32 radical prostatectomy samples, confirms upregulation of PRDX-3 in tumours compared with benign and also shows the upregulation of PRDX-3 in PIN, suggesting that its overexpression may be an early event in tumourigenesis. Previous studies have suggested that 33% of adjacent normal prostate tissue may demonstrate some raised expression of PRDX-3, consistent with a field effect seen in other solid tumours (Basu et al, 2011). Although the benign on our TMA was taken from the same patient, whenever possible, it was not taken directly from the adjacent tumour. Any field effect is likely to raise expression in the benign cohort, making any differences in expression between benign and tumour even more significant. Our results are also consistent with a previous study showing a negative correlation between PRDX-3 overexpression and TMPRSS–ERG fusion status in PCa (Ummanni et al, 2012).
The localisation and of the PRDX proteins is varied with PRDX-3 predominantly localised to the mitochondria and, to a lesser extent, the cell surface (Araki et al, 1999; Liu et al, 2005; Whitaker et al, 2007) (Figure 2). The localisation of the mitochondrial proteins with mitochondria and the cellular membrane has previously been reported for p32 gC1qR, a protein with no defined function, this is, thought to act as a generic chaperone (Fogal et al, 2008). Our group has previously reported downregulation of cell surface and total cellular PRDX-3 in response to androgen treatment (Whitaker et al, 2007). We have confirmed this androgen regulation of PRDX-3 using gene expression data showing a decrease in the PRDX-3 RNA expression in response to R1881 treatment (Figure 2C). This reduction in PRDX-3 corresponds to a loss of cell surface PRDX-3, consistent with our previous results (Figure 2B) (Whitaker et al, 2007). The localisation of the mitochondrial proteins with mitochondria and the cellular membrane has previously been reported for p32 gC1qR, a protein of unknown function that is being studied for potential therapeutic utility.
The protective role of PRDX-3 in oxidative stress has previously been shown in the cardiovascular system (Araki et al, 1999) and in response to pro-apoptotic drugs, such as auranofin and isothiocyanates in tumour cells (Brown et al, 2008; Cox et al, 2008a). We report that PRDX-3 down regulated by R1881 treatment despite reports that androgen treatment increases fatty-acid synthesis and ROS production (Lin et al, 2010). PRDX-3 is highly upregulated in the antiandrogen-resistant cell lines, indicating that it may have an antiapoptotic function in these cells and promote their survival (Figure 3A). Interestingly, upregulation occurs at the protein but not the mRNA level, suggesting that regulation of PRDX-3 occurs at the translation or protein level and this is consistent with an increase in the oxidised form of PRDX-3 seen after antiandrogen treatment (Figures 3A, B and 4A). Our data suggest that the stabilisation of PRDX-3 via oxidation and its subsequent, slower, recycling back to the reduced and active form may be an additional layer of regulation of peroxiredoxin expression levels in prostate cells. Upregulation of PRDX-3 was relatively rapid, occurring after only 3 days bicalutamide treatment, suggesting that raised PRDX-3 may be part of the mechanism leading to antiandrogen resistance rather than an increase occurring as a result of resistance (Figure 4A). The accompanying increase in oxidised PRDX-3 supports the hypothesis that higher levels of oxidative stress exist in cells treated with antiandrogen. Although PRDX-3 oxidation has been linked to apoptosis via Bax/Bak, it may also represent the mechanism by which PRDX-3 can be stabilized and recycled by thioredoxin-2 in resistant cells (Wood et al, 2003; Cox et al, 2008a).
PRDX-3 is known to catalyse the reduction of ROS, including H2O2, which are produced as a result of cellular metabolism, for example, ATP synthesis. A major site of ROS production is the mitochondria, organelles which can also regulate apoptosis (Rustin and Kroemer, 2007). Bicalutamide-resistant cells that contain higher levels of PRDX-3 also overexpress COX IV and possessed four- to six-fold higher citrate synthase activity (Figure 4A). Citrate synthase is the rate limiting first enzyme in the TCA cycle and represents the metabolic activity via mitochondrial oxidative phosphorylation occurring within a cell. We hypothesise that a subset of prostate cells with greater mitochondrial number can overcome the oxidative stress and pro-apoptotic signals caused by antiandrogen treatment due to higher metabolic capacity and an increased resistance to oxidative stress due to increased PRDX-3 levels. Consistent with this, others have shown that mutations in mitochondrial DNA are essential for conferring androgen independence in the LNCaP derivative cell lines C4-2 (Higuchi et al, 2006).
Concomitant with the raised levels of PRDX-3, antiandrogen-resistant cells show an increased resistance to H2O2, particularly at low concentrations, where PRDX proteins are known to be most efficient (Lee et al, 2007; Cox et al, 2009). Treatment of bicalutamide-resistant cells with H2O2 failed to induce the pro-apoptotic signals seen in the parental cell lines (Figure 4C). This effect was reversed by knocking down PRDX-3 in LNCaP-BIC cells. Conversely knocking down PRDX-3 in parental LNCaP cells makes them more susceptible to H2O2-mediated apoptosis as previously noted following TNF-α treatment (Cox et al, 2008b). This confirms a previous report that PRDX-3 is responsible for increased resistance to H2O2, and suggests that PRDX-3 overexpression may have a role in antiandrogen resistance (Chen et al, 2008). We suggest that metabolic adaptation and increase in the mitochondrial number in prostate cells may result in resistance to the oxidative stress and pro-apoptotic signals initiated by hormone therapy, as seen in neuronal cells and breast cancer xenografts (Besada et al, 2006; Busija et al, 2008; De Simoni et al, 2008). ROS regulation is a novel mechanism leading to anti-androgen resistance which could be exploited for as therapeutic targets for small-molecule inhibitors or radiotherapy (Trachootham et al, 2009; Zhang et al, 2009).
Change history
20 August 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ambruso DR, Ellison MA, Thurman GW, Leto TL (2012) Peroxiredoxin 6 translocates to the plasma membrane during neutrophil activation and is required for optimal NADPH oxidase activity. Biochim Biophys Acta 1823 (2): 306–315.
Araki M, Nanri H, Ejima K, Murasato Y, Fujiwara T, Nakashima Y, Ikeda M (1999) Antioxidant function of the mitochondrial protein SP-22 in the cardiovascular system. J Biol Chem 274 (4): 2271–2278.
Aran M, Ferrero DS, Pagano E, Wolosiuk RA (2009) Typical 2-Cys peroxiredoxins--modulation by covalent transformations and noncovalent interactions. FEBS J 276 (9): 2478–2493.
Basu A, Banerjee H, Rojas H, Martinez SR, Roy S, Jia Z, Lilly MB, De Leon M, Casiano CA (2011) Differential expression of peroxiredoxins in prostate cancer: consistent upregulation of PRDX3 and PRDX4. The Prostate 71 (7): 755–765.
Besada V, Diaz M, Becker M, Ramos Y, Castellanos-Serra L, Fichtner I (2006) Proteomics of xenografted human breast cancer indicates novel targets related to tamoxifen resistance. Proteomics 6 (3): 1038–1048.
Bevan CL (2005) Androgen receptor in prostate cancer: cause or cure? Trendsendocrinol metab 16 (9): 395–397.
Brown KK, Eriksson SE, Arner ES, Hampton MB (2008) Mitochondrial peroxiredoxin 3 is rapidly oxidised in cells treated with isothiocyanates. Free Radic Biol Med 45 (4): 494–502.
Busija DW, Gaspar T, Domoki F, Katakam PV, Bari F (2008) Mitochondrial-mediated suppression of ROS production upon exposure of neurons to lethal stress: mitochondrial targeted preconditioning. Adv Drug Deliv Rev 60 (13-14): 1471–1477.
Cairns JM, Dunning MJ, Ritchie ME, Russell R, Lynch AG (2008) BASH: a tool for managing BeadArray spatial artefacts. Bioinformatics 24 (24): 2921–2922.
Cha MK, Suh KH, Kim IH (2009) Overexpression of peroxiredoxin I and thioredoxin1 in human breast carcinoma. J Exp Clin Cancer Res 28: 93.
Chang KP, Yu JS, Chien KY, Lee CW, Liang Y, Liao CT, Yen TC, Lee LY, Huang LL, Liu SC, Chang YS, Chi LM (2011) Identification of PRDX4 and P4HA2 as metastasis-associated proteins in oral cavity squamous cell carcinoma by comparative tissue proteomics of microdissected specimens using iTRAQ technology. J Proteome Res 10 (11): 4935–4947.
Chang XZ, Li DQ, Hou YF, Wu J, Lu JS, Di GH, Jin W, Ou ZL, Shen ZZ, Shao ZM (2007) Identification of the functional role of peroxiredoxin 6 in the progression of breast cancer. Breast Cancer Res 9 (6): R76.
Chen L, Na R, Gu M, Salmon AB, Liu Y, Liang H, Qi W, Van Remmen H, Richardson A, Ran Q (2008) Reduction of mitochondrial H2O2 by overexpressing peroxiredoxin 3 improves glucose tolerance in mice. Aging Cell 7 (6): 866–878.
Chhipa RR, Lee KS, Onate S, Wu Y, Ip C (2009) Prx1 enhances androgen receptor function in prostate cancer cells by increasing receptor affinity to dihydrotestosterone. Mol Cancer Res 7 (9): 1543–1552.
Cho YE, Singh TS, Lee HC, Moon PG, Lee JE, Lee MH, Choi EC, Chen YJ, Kim SH, Baek MC (2012) In-depth identification of pathways related to cisplatin-induced hepatotoxicity through an integrative method based on an informatics-assisted label-free protein quantitation and microarray gene expression approach. Mol Cell Proteomics: MCP 11 (1): M111 010884.
Choi JH, Kim TN, Kim S, Baek SH, Kim JH, Lee SR, Kim JR (2002) Overexpression of mitochondrial thioredoxin reductase and peroxiredoxin III in hepatocellular carcinomas. Anticancer Res 22 (6A): 3331–3335.
Chua PJ, Lee EH, Yu Y, Yip GW, Tan PH, Bay BH (2010) Silencing the Peroxiredoxin III gene inhibits cell proliferation in breast cancer. Int J Oncol 36 (2): 359–364.
Cox AG, Brown KK, Arner ES, Hampton MB (2008a) The thioredoxin reductase inhibitor auranofin triggers apoptosis through a Bax/Bak-dependent process that involves peroxiredoxin 3 oxidation. Biochem Pharmacol 76 (9): 1097–1109.
Cox AG, Peskin AV, Paton LN, Winterbourn CC, Hampton MB (2009) Redox potential and peroxide reactivity of human peroxiredoxin 3. Biochemistry 48 (27): 6495–6501.
Cox AG, Pullar JM, Hughes G, Ledgerwood EC, Hampton MB (2008b) Oxidation of mitochondrial peroxiredoxin 3 during the initiation of receptor-mediated apoptosis. Free Rad Biol Med 44 (6): 1001–1009.
De Simoni S, Goemaere J, Knoops B (2008) Silencing of peroxiredoxin 3 and peroxiredoxin 5 reveals the role of mitochondrial peroxiredoxins in the protection of human neuroblastoma SH-SY5Y cells toward MPP+. Neurosci Lett 433 (3): 219–224.
Dittmann LM, Danner A, Gronych J, Wolter M, Stuhler K, Grzendowski M, Becker N, Bageritz J, Goidts V, Toedt G, Felsberg J, Sabel MC, Barbus S, Reifenberger G, Lichter P, Tews B (2011) Downregulation of PRDX1 by promoter hypermethylation is frequent in 1p/19q-deleted oligodendroglial tumours and increases radio- and chemosensitivity of Hs683 glioma cells in vitro. Oncogene 31: 3409–3418.
Dunning MJ, Smith ML, Ritchie ME, Tavare S (2007) Beadarray: R classes and methods for Illumina bead-based data. Bioinformatics 23 (16): 2183–2184.
Fogal V, Zhang L, Krajewski S, Ruoslahti E (2008) Mitochondrial/cell-surface protein p32/gC1qR as a molecular target in tumor cells and tumor stroma. Cancer Res 68 (17): 7210–7218.
Friedlander TW, Ryan CJ (2012) Targeting the androgen receptor. Urol Clin North Am 39 (4): 453–464.
Fujii J, Ikeda Y (2002) Advances in our understanding of peroxiredoxin, a multifunctional, mammalian redox protein. Redox Rep 7 (3): 123–130.
Gao MC, Jia XD, Wu QF, Cheng Y, Chen FR, Zhang J (2011) Silencing Prx1 and/or Prx5 sensitizes human esophageal cancer cells to ionizing radiation and increases apoptosis via intracellular ROS accumulation. Acta Pharmacol Sin 32 (4): 528–536.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5 (10): R80.
Giorgio M, Trinei M, Migliaccio E, Pelicci PG (2007) Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev 8 (9): 722–728.
Hall A, Karplus PA, Poole LB (2009) Typical 2-Cys peroxiredoxins--structures, mechanisms and functions. FEBS J 276 (9): 2469–2477.
Hanschmann EM, Lonn ME, Schutte LD, Funke M, Godoy JR, Eitner S, Hudemann C, Lillig CH (2010) Both thioredoxin 2 and glutaredoxin 2 contribute to the reduction of the mitochondrial 2-Cys peroxiredoxin Prx3. J Biol Chem 285 (52): 40699–40705.
He HC, Zhu JG, Chen XB, Chen SM, Han ZD, Dai QS, Ling XH, Fu X, Lin ZY, Deng YH, Qin GQ, Cai C, Chen JH, Zhong WD (2012) MicroRNA-23b downregulates peroxiredoxin III in human prostate cancer. FEBS Lett 586 (16): 2451–2458.
Higuchi M, Kudo T, Suzuki S, Evans TT, Sasaki R, Wada Y, Shirakawa T, Sawyer JR, Gotoh A (2006) Mitochondrial DNA determines androgen dependence in prostate cancer cell lines. Oncogene 25 (10): 1437–1445.
Hobisch A, Fritzer A, Comuzzi B, Fiechtl M, Malinowska K, Steiner H, Bartsch G, Culig Z (2006) The androgen receptor pathway is by-passed in prostate cancer cells generated after prolonged treatment with bicalutamide. Prostate 66 (4): 413–420.
Hoskins ER, Hood BL, Sun M, Krivak TC, Edwards RP, Conrads TP (2011) Proteomic analysis of ovarian cancer proximal fluids: validation of elevated peroxiredoxin 1 in patient peripheral circulation. PLoS One 6 (9): e25056.
Karihtala P, Kauppila S, Soini Y, Arja Jukkola V (2011) Oxidative stress and counteracting mechanisms in hormone receptor positive, triple-negative and basal-like breast carcinomas. BMC Cancer 11: 262.
Karihtala P, Mantyniemi A, Kang SW, Kinnula VL, Soini Y (2003) Peroxiredoxins in breast carcinoma. Clin Cancer Res 9 (9): 3418–3424.
Kikuta K, Tochigi N, Saito S, Shimoda T, Morioka H, Toyama Y, Hosono A, Suehara Y, Beppu Y, Kawai A, Hirohashi S, Kondo T (2010) Peroxiredoxin 2 as a chemotherapy responsiveness biomarker candidate in osteosarcoma revealed by proteomics. Proteomics Clin Appl 4 (5): 560–567.
Kim JH, Bogner PN, Baek SH, Ramnath N, Liang P, Kim HR, Andrews C, Park YM (2008) Up-regulation of peroxiredoxin 1 in lung cancer and its implication as a prognostic and therapeutic target. Clin Cancer Res 14 (8): 2326–2333.
Kim K, Yu M, Han S, Oh I, Choi YJ, Kim S, Yoon K, Jung M, Choe W (2009) Expression of human peroxiredoxin isoforms in response to cervical carcinogenesis. Oncol Rep 21 (6): 1391–1396.
Kinnula VL, Lehtonen S, Sormunen R, Kaarteenaho-Wiik R, Kang SW, Rhee SG, Soini Y (2002) Overexpression of peroxiredoxins I, II, III, V, and VI in malignant mesothelioma. J Pathol 196 (3): 316–323.
Kuil CW, Mulder E (1994) Mechanism of antiandrogen action: conformational changes of the receptor. Mol Cell Endocrinol 102 (1-2): R1–R5.
Kumar V, Kitaeff N, Hampton MB, Cannell MB, Winterbourn CC (2009) Reversible oxidation of mitochondrial peroxiredoxin 3 in mouse heart subjected to ischemia and reperfusion. FEBS Lett 583 (6): 997–1000.
Lee KW, Lee DJ, Lee JY, Kang DH, Kwon J, Kang SW (2011) Peroxiredoxin II restrains DNA damage-induced death in cancer cells by positively regulating JNK-dependent DNA repair. J Biol Chem 286 (10): 8394–8404.
Lee W, Wells T, Kantorow M (2007) Localization and H2O2-specific induction of PRDX3 in the eye lens. Mol Vis 13: 1469–1474.
Lehtonen ST, Svensk AM, Soini Y, Paakko P, Hirvikoski P, Kang SW, Saily M, Kinnula VL (2004) Peroxiredoxins, a novel protein family in lung cancer. Int J Cancer 111 (4): 514–521.
Lenaz G (2012) Mitochondria and reactive oxygen species. Which role in physiology and pathology? Adv Exp Med Biol 942: 93–136.
Leyens G, Donnay I, Knoops B (2003) Cloning of bovine peroxiredoxins-gene expression in bovine tissues and amino acid sequence comparison with rat, mouse and primate peroxiredoxins. Comp BiochemPhysiol 136 (4): 943–955.
Li KK, Pang JC, Lau KM, Zhou L, Mao Y, Wang Y, Poon WS, Ng HK (2012) MiR-383 is downregulated in medulloblastoma and targets Peroxiredoxin 3 (PRDX3). Brain Pathol 23: 413–425.
Lin H, Lu JP, Laflamme P, Qiao S, Shayegan B, Bryskin I, Monardo L, Wilson BC, Singh G, Pinthus JH (2010) Inter-related in vitro effects of androgens, fatty acids and oxidative stress in prostate cancer: a mechanistic model supporting prevention strategies. Int J Oncol 37 (4): 761–766.
Lin JF, Xu J, Tian HY, Gao X, Chen QX, Gu Q, Xu GJ, Song JD, Zhao FK (2007) Identification of candidate prostate cancer biomarkers in prostate needle biopsy specimens using proteomic analysis. Int J Cancer 121 (12): 2596–2605.
Liu L, Yang C, Yuan J, Chen X, Xu J, Wei Y, Yang J, Lin G, Yu L (2005) RPK118, a PX domain-containing protein, interacts with peroxiredoxin-3 through pseudo-kinase domains. Mol Cells 19 (1): 39–45.
Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, Warren A, Scott H, Madhu B, Sharma N, Bon H, Zecchini V, Smith DM, Denicola GM, Mathews N, Osborne M, Hadfield J, Macarthur S, Adryan B, Lyons SK, Brindle KM, Griffiths J, Gleave ME, Rennie PS, Neal DE, Mills IG (2011) The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J 30 (13): 2719–2733.
Maxwell GL, Hood BL, Day R, Chandran U, Kirchner D, Kolli VS, Bateman NW, Allard J, Miller C, Sun M, Flint MS, Zahn C, Oliver J, Banerjee S, Litzi T, Parwani A, Sandburg G, Rose S, Becich MJ, Berchuck A, Kohn E, Risinger JI, Conrads TP (2011) Proteomic analysis of stage I endometrial cancer tissue: identification of proteins associated with oxidative processes and inflammation. Gynecol Oncol 121 (3): 586–594.
Miki H, Funato Y (2012) Regulation of intracellular signalling through cysteine oxidation by reactive oxygen species. J Biochem 151 (3): 255–261.
Noh DY, Ahn SJ, Lee RA, Kim SW, Park IA, Chae HZ (2001) Overexpression of peroxiredoxin in human breast cancer. Anticancer Res 21 (3B): 2085–2090.
Pak JH, Choi WH, Lee HM, Joo WD, Kim JH, Kim YT, Kim YM, Nam JH (2011) Peroxiredoxin 6 overexpression attenuates cisplatin-induced apoptosis in human ovarian cancer cells. Cancer Invest 29 (1): 21–28.
Palande KK, Beekman R, van der Meeren LE, Beverloo HB, Valk PJ, Touw IP (2011) The antioxidant protein peroxiredoxin 4 is epigenetically down regulated in acute promyelocytic leukemia. PLoS ONE 6 (1): e16340.
Park JH, Kim YS, Lee HL, Shim JY, Lee KS, Oh YJ, Shin SS, Choi YH, Park KJ, Park RW, Hwang SC (2006) Expression of peroxiredoxin and thioredoxin in human lung cancer and paired normal lung. Respirology 11 (3): 269–275.
Park JY, Kim SA, Chung JW, Bang S, Park SW, Paik YK, Song SY (2011) Proteomic analysis of pancreatic juice for the identification of biomarkers of pancreatic cancer. J Cancer Res Clin Oncol 137 (8): 1229–1238.
Park SY, Yu X, Ip C, Mohler JL, Bogner PN, Park YM (2007) Peroxiredoxin 1 interacts with androgen receptor and enhances its transactivation. Cancer Res 67 (19): 9294–9303.
Pylvas M, Puistola U, Kauppila S, Soini Y, Karihtala P (2010) Oxidative stress-induced antioxidant enzyme expression is an early phenomenon in ovarian carcinogenesis. Eur J Cancer 46 (9): 1661–1667.
Quan C, Cha EJ, Lee HL, Han KH, Lee KM, Kim WJ (2006) Enhanced expression of peroxiredoxin I and VI correlates with development, recurrence and progression of human bladder cancer. J Urol 175 (4): 1512–1516.
Ray PD, Huang BW, Tsuji Y (2012) Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24 (5): 981–990.
Ren L, Yang HY, Choi HI, Chung KJ, Yang U, Lee IK, Kim HJ, Lee DS, Park BJ, Lee TH (2011) The role of peroxiredoxin V in (-)-epigallocatechin 3-gallate-induced multiple myeloma cell death. Oncol Res 19 (8-9): 391–398.
Ruckenstuhl C, Buttner S, Carmona-Gutierrez D, Eisenberg T, Kroemer G, Sigrist SJ, Frohlich KU, Madeo F (2009) The Warburg effect suppresses oxidative stress induced apoptosis in a yeast model for cancer. PLoS One 4 (2): e4592.
Rustin P, Kroemer G (2007) Mitochondria and cancer. Ernst Schering Found Sym Proc 4: 1–21.
Shen C, Nathan C (2002) Nonredundant antioxidant defense by multiple two-cysteine peroxiredoxins in human prostate cancer cells. Mol Med 8 (2): 95–102.
Shiota M, Yokomizo A, Kashiwagi E, Takeuchi A, Fujimoto N, Uchiumi T, Naito S (2011) Peroxiredoxin 2 in the nucleus and cytoplasm distinctly regulates androgen receptor activity in prostate cancer cells. Free Rad Biol Med 51 (1): 78–87.
Smyth GK (2005) Limma: linear models for microarray data in R. Springer: New York.
Sofiadis A, Becker S, Hellman U, Hultin-Rosenberg L, Dinets A, Hulchiy M, Zedenius J, Wallin G, Foukakis T, Hoog A, Auer G, Lehtio J, Larsson C (2012) Proteomic profiling of follicular and papillary thyroid tumors. Eur J Endocrinol 166: 657–667.
Svendsen A, Verhoeff JJ, Immervoll H, Brogger JC, Kmiecik J, Poli A, Netland IA, Prestegarden L, Planaguma J, Torsvik A, Kjersem AB, Sakariassen PO, Heggdal JI, Van Furth WR, Bjerkvig R, Lund-Johansen M, Enger PO, Felsberg J, Brons NH, Tronstad KJ, Waha A, Chekenya M (2011) Expression of the progenitor marker NG2/CSPG4 predicts poor survival and resistance to ionising radiation in glioblastoma. Acta Neuropathol 122 (4): 495–510.
Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Theodore C, James ND, Turesson I, Rosenthal MA, Eisenberger MA (2004) Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 351 (15): 1502–1512.
Thongwatchara P, Promwikorn W, Srisomsap C, Chokchaichamnankit D, Boonyaphiphat P, Thongsuksai P (2011) Differential protein expression in primary breast cancer and matched axillary node metastasis. Oncol Rep 26 (1): 185–191.
Trachootham D, Alexandre J, Huang P (2009) Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 8 (7): 579–591.
Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P (2008) Redox regulation of cell survival. Antioxid Redox Signal 10 (8): 1343–1374.
Ummanni R, Barreto F, Venz S, Scharf C, Barett C, Mannsperger HA, Brase JC, Kuner R, Schlomm T, Sauter G, Sultmann H, Korf U, Bokemeyer C, Walther R, Brummendorf TH, Balabanov S (2012) Peroxiredoxins 3 and 4 are overexpressed in prostate cancer tissue and affect the proliferation of prostate cancer cells in vitro. J Proteome Res 11 (4): 2452–2466.
Vias M, Burtt G, Culig Z, Veerakumarasivam A, Neal DE, Mills IG (2006) A role for neurotensin in bicalutamide resistant prostate cancer cells. Prostate 67 (2): 190–202.
Vivas-Mejia PE, Ozpolat B, Chen X, Lopez-Berestein G (2009) Downregulation of the c-MYC target gene, peroxiredoxin III, contributes to arsenic trioxide-induced apoptosis in acute promyelocytic leukemia. Int J Cancer 125 (2): 264–275.
Whitaker HC, Girling J, Warren AY, Leung H, Mills IG, Neal DE (2008) Alterations in beta-catenin expression and localization in prostate cancer. Prostate 68 (11): 1196–1205.
Whitaker HC, Kote-Jarai Z, Ross-Adams H, Warren AY, Burge J, George A, Bancroft E, Jhavar S, Leongamornlert D, Tymrakiewicz M, Saunders E, Page E, Mitra A, Mitchell G, Lindeman GJ, Evans DG, Blanco I, Mercer C, Rubinstein WS, Clowes V, Douglas F, Hodgson S, Walker L, Donaldson A, Izatt L, Dorkins H, Male A, Tucker K, Stapleton A, Lam J, Kirk J, Lilja H, Easton D, Cooper C, Eeles R, Neal DE (2010) The rs10993994 risk allele for prostate cancer results in clinically relevant changes in microseminoprotein-beta expression in tissue and urine. PLoS One 5 (10): e13363.
Whitaker HC, Stanbury DP, Brinham C, Girling J, Hanrahan S, Totty N, Neal DE (2007) Labeling and identification of LNCaP cell surface proteins: a pilot study. Prostate 67 (9): 943–954.
Wonsey DR, Zeller KI, Dang CV (2002) The c-Myc target gene PRDX3 is required for mitochondrial homeostasis and neoplastic transformation. Proc Natl Acad Sci USA 99 (10): 6649–6654.
Wood ZA, Schroder E, Robin Harris J, Poole LB (2003) Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci 28 (1): 32–40.
Woolston CM, Storr SJ, Ellis IO, Morgan DA, Martin SG (2011) Expression of thioredoxin system and related peroxiredoxin proteins is associated with clinical outcome in radiotherapy treated early stage breast cancer. Radiother Oncol 100 (2): 308–313.
Zhang B, Wang K, He G, Guan X, Liu B, Liu Y, Bai Y (2012) Polymorphisms of peroxiredoxin 1, 2 and 6 are not associated with esophageal cancer. J Cancer Res Clin Oncol 138: 621–626.
Zhang B, Wang Y, Su Y (2009) Peroxiredoxins, a novel target in cancer radiotherapy. Cancer Lett 286 (2): 154–160.
Zhang J, Wang K, Liu SS, Dai L, Zhang JY (2011) Using proteomic approach to identify tumor-associated proteins as biomarkers in human esophageal squamous cell carcinoma. J Proteome Res 10 (6): 2863–2872.
Zhiyu W, Yue C, Neng W, Mei WD, Wei LY, Feng H, Gang SJ, De PoY, Yuan GX, Jian-Ping C (2012) Dioscin induces cancer cell apoptosis through elevated oxidative stress mediated by downregulation of peroxiredoxins. Cancer Biol Ther 13 (3): 138–147.
Acknowledgements
We acknowledge the support of The University of Cambridge, Cancer Research UK and Hutchison Whampoa Limited. We also acknowledge the support of the National Institute for Health Research (NIHR) which funds the Cambridge Bio-medical Research Centre, Cambridge UK. We would also like to acknowledge the support of the NCRI (ProMPT) which has funded tissue collections in Cambridge. We are grateful to Zoran Culig (Innesbruck) for the antiandrogen-resistant cell lines. We also thank Jason Flory (IT and Scientific Computing, CRUK Cambridge Institute) for the help and advice with publishing tools. This work was funded by Prostate Cancer UK, Cancer Research UK and the Medical Research Council.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Whitaker, H., Patel, D., Howat, W. et al. Peroxiredoxin-3 is overexpressed in prostate cancer and promotes cancer cell survival by protecting cells from oxidative stress. Br J Cancer 109, 983–993 (2013). https://doi.org/10.1038/bjc.2013.396
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.396
