
Abstract
NAD[P]H:quinone oxidoreductase 1 (NQO1) regulates cell fate decisions in response to stress. Oxidative stress supports cancer maintenance and progression. Previously we showed that knockdown of NQO1 (NQO1low) prostate cancer cells upregulate pro-inflammatory cytokines and survival under hormone-deprived conditions. Here, we tested the ability of NQO1low cells to form tumors. We found NQO1low cells form aggressive tumors compared with NQO1high cells. Biopsy specimens and circulating tumor cells showed biochemical recurrent prostate cancer was associated with low NQO1. NQO1 silencing was sufficient to induce SMAD-mediated TGFβ signaling and mesenchymal markers. TGFβ treatment decreased NQO1 levels and induced molecular changes similar to NQO1 knockdown cells. Functionally, NQO1 depletion increased migration and sensitivity to oxidative stress. Collectively, this work reveals a possible new gatekeeper role for NQO1 in counteracting cellular plasticity in prostate cancer cells. Further, combining NQO1 with TGFβ signaling molecules may serve as a better signature to predict biochemical recurrence.
Similar content being viewed by others

Introduction
Cancer cells are under increased oxidative stress, presumably associated with uncontrolled growth, metastasis, and response to therapy. Cancer cells have an intrinsic antioxidant defense system through a battery of antioxidant enzymes to adapt to increased oxidative stress1. NAD[P]H:quinone oxidoreductase 1 (NQO1) is a key component of the antioxidant defense system. NQO1 is a cytosolic enzyme that detoxifies quinones and protects cells against oxidative stress. Several studies have revealed protective roles for NQO1 that apparently are unrelated to its enzymatic activities. For example, NQO1 directly binds to the tumor suppressor p53 protein and stabilizes it against proteasomal degradation ultimately making cell fate decisions in response to endogenous and exogenous stress2,3.
Oxidative stress is recognized as an important contributor to the transition of epithelial cells to mesenchymal phenotype (EMT), a reversible program that enables metastasis4. This switch in cell differentiation and behavior is mediated by key transcription factors, including SNAIL, zinc-finger E-box-binding (ZEB), and basic helix-loop-helix transcription factors5. EMT is characterized by upregulation of mesenchymal-associated genes, such as N-cadherin, vimentin and fibronectin, and decreased expression of epithelial-associated genes such as E-cadherin5. These changes in cell plasticity are associated with aggressive cell behavior including migration, invasion, tumor cell survival, stemness, resistance to radiation and chemotherapy in various cancer types including prostate cancer6. The reversible nature of the EMT process ensures that mesenchymal cells undergo differentiation back to epithelial phenotype (mesenchymal-epithelial transition; MET). Therefore, these transient molecular changes are initiated and controlled by signaling pathways that respond to extracellular cues such as oxidative stress and not by permanent alteration5,7. Transforming growth factor-β (TGFβ) family plays a leading role in this signaling process.
TGFβ is a pleotropic cytokine, which upon activation causes autocrine and paracrine effects on tumor cells and the tumor microenvironment, thus regulating a variety of cellular processes, including angiogenesis, apoptosis, migration, EMT, and metastasis8,9. Several studies have suggested that in early-stage cancers, TGFβ signaling may be growth suppressive, while in advanced stages, TGFβ promotes invasion and metastasis10,11. TGFβ signaling in prostate cancer cells increases the expression of numerous genes associated with the development of bone metastases and TGFβ inhibition effectively reduces prostate cancer bone metastases12. Recent studies highlight the immunosuppressive role of TGFβ leading to immune evasion therefore it can be targeted to improve cancer immunotherapy13,14. TGFβ receptors initiate downstream signaling through either SMAD-mediated canonical signaling or SMAD-independent non-canonical signaling. The canonical signaling pathway involves phosphorylation of SMAD2 or SMAD3 with SMAD4 and nuclear translocation to regulate gene expression with the help of transcriptional coactivators or corepressors11.
Prostate cancer is one of the most commonly diagnosed malignancy accounting for almost 1 in 5 cancer diagnoses among men in the United States15. Oxidative stress, a hallmark of prostate cancer is correlated with tumor metastasis and therapeutic resistance in primary tumors16,17,18. We had reported that NQO1 attenuation fueled pro-inflammatory signaling and promoted androgen-independent prostate cancer cell survival19. Interestingly, Kurfurstova et al. observed a focal loss of NQO1 along with loss of PTEN in advanced prostate cancer lesions suggesting NQO1 may have a tumor suppressive role and its loss may facilitate tumor progression20. In contrast, recent reports suggest a pro-tumorigenic role of NQO1 in breast and colon cancers since high expression was implicated in disease progression and metastasis21,22. Despite these paradoxical but critical observations regarding NQO1 in various cancers, and the contribution of oxidative stress and inflammation to prostate cancer development and progression, the clinicopathological significance of NQO1 in prostate cancer and the molecular mechanisms underlying NQO1 regulation have not been fully elucidated. Our goal in this work was to understand how NQO1 contributes to prostate carcinogenesis16,23.
Here we show that indeed NQO1 inhibition enhanced prostate tumorigenesis in an orthotopic model. NQO1 message and protein is reduced in advanced prostate cancer, is associated with survival in multiple cohorts and biochemical recurrence (BCR) in circulating tumor cells (CTC) obtained from prostate cancer patients. We also show that suppression of NQO1 induced EMT through TGFβ signaling activation. TGFβ treatment of prostate cancer cells decreased NQO1 levels and mimicked molecular changes of NQO1 knockdown cells. Together our results provide evidence that NQO1 acts as a guardian to protect prostate cancer cells from undergoing TGFβ-mediated EMT changes that are associated with advanced disease progression.
Results
NQO1 knockdown accelerates prostate tumorigenesis
Our previous work showed that NQO1low cells upregulated inflammatory signaling19. To determine the impact of low NQO1 on prostate tumorigenesis, stably silenced NQO1 (shNQO1) cells and non-target control (NTC) were implanted into the prostate of athymic nude mice, followed by analysis of tumor growth. At the time of euthanasia, five of seven animals with shNQO1 cells developed tumors, while there was no incidence of tumor in mice (0/8) implanted with NTC cells (Fig. 1a). Prostates from animals bearing shNQO1 tumors weighed more than those from animals implanted with NTC cells (Fig. 1b). Pathological analyses showed that five of seven animals in the shNQO1 group developed poorly differentiated carcinoma (PDC), while none of the animals in the NTC group developed PDC (Fig. 1c and Table 1). All animals in the NTC group developed high-grade intraepithelial neoplasia (HGPIN) which we categorized into 0–3 grades based on their histology (Supplementary Table 1). We then analyzed our microarray data set of shNQO1 and NTC cells (GSE58336)19. Using DAVID (Database for Annotation, Visualization and Integrated Discovery) web tool, we identified several pathways associated with tumorigenesis including functions related to wounding, chemotaxis, and migration (Fig. 1d, e). To our knowledge, this study shows for the first time that NQO1 knockdown enhances prostate tumor development in vivo.

a Prostate and seminal vesicles showing tumor burden in animals with orthotopic injection of NTC and shNQ cells. b Weights of prostate and prostate + tumor. c Representative images of H&E-stained sections from the prostate and tumor. Scale bars, 500 µm (low x) and 50 µm (high x). d, e Genome wide expression changes between NTC and shNQ cells were analyzed for pathways and functions using DAVID web tool. DAVID-generated modified Fisher’s exact test p-values of selected terms are represented in log-10 scale.
Reduced NQO1 is associated with advanced prostate cancer
Analysis of publicly available datasets for NQO1 expression in surgical specimens showed significantly lower expression in metastatic tumors (liver, lymph node, lung, adrenal; (P < 0.0001 with unpaired t-test) compared with primary prostate tumors (Fig. 2a)24. Expression data from GSE35988 showed significant association (P < 0.0001 with unpaired t-test) between low expression of NQO1 and metastatic prostate cancer25 (Fig. 2b). NQO1 expression decreased consistently in metastatic prostate cancer samples in multiple independent studies26,27,28,29,30 (Supplementary Fig. 1). To determine whether NQO1 predicts prostate cancer patient outcomes, we investigated the correlation and prognostic implication of NQO1 in two independent cohorts GSE70769 and GSE40272 using PROGgeneV231. Consistent with the expression of low NQO1 and tumor metastasis in multiple cohorts, a strong association was observed between NQO1 mRNA expression and prognosis of prostate cancer patients. Patients with low NQO1 expression had poorer relapse free survival (Fig. 2c and Supplementary Fig. 2).

a NQO1 expression in primary prostate tumors and in samples from distant metastases (Chandran dataset). Expression level is presented as boxplot (P < 0.0001 with unpaired t-test). b Scatter plot analysis of NQO1 expression in tumor and metastasis (GSE35988). c Kaplan–Meier analysis of recurrence-free survival (RFS) was compared between high and low NQO1 transcript levels using median gene expression value as a bifurcating point. RFS plot of 91 prostate cancer patients from GSE70769. d Representative photographs of immunohistochemical detection of NQO1 in a tumor microarray with a cohort representing benign, PIN and carcinoma lesions obtained at the time of prostatectomy (n = 150). Scale bars, 100 µm (e). Kaplan–Meier analysis of recurrence-free survival in the TMA (from above) based on low (0/1+) and high (2+/3+) immunostaining (P = 0.329 with log-rank analysis). f Composition of the 23 BCR patients from the TMA samples with a NQO1 change. Graphs showing cumulative IHC staining scores from benign and cancer sites from each patient. g t-Distributed Stochastic Neighbor Embedding (t-SNE) plot of unbiased clustering of 136 CTCs from patients (n = 9) based on NQO1 and TGF-β pathway scores, and identification of four groups in different colors. h NQO1 expressed as percentage in each group. Initial diagnosis (ID) and biochemical recurrence (BCR) ratio in different groups (p values calculated by Chi-square test).
We then used a set of tissue microarray (TMA) consisting of a cohort of 150 radical prostatectomy specimens. NQO1 was detectable mainly in the cytoplasmic compartment of epithelial cells from benign, high-grade prostatic intraepithelial neoplasia (PIN), and infiltrating adenocarcinoma. In multiple cases, there was a notable decrease in NQO1 level (0 to 1+) in carcinoma compared with PIN from the same patient that showed strong staining (2+ to 3+) (Fig. 2d). Low levels (0 to 1+) of NQO1 were seen in 49% of carcinomas versus 30% and 37% of benign and PIN tissues, respectively (P < 0.05) (Supplementary Table 2). In particular, 22 (56%) of 39 advanced tumors (i.e., pT3-4 and/or pN1) showed low NQO1 expression. However, analyses of clinicopathological data of this cohort showed no statistically significant correlations between the level of NQO1 protein and Gleason score (GS), pathologic stage (pT), and lymph node metastasis (pN) (Supplementary Table 3). There was a trend of an association between low NQO1 protein level and decreased recurrence-free survival, however the difference was not significant (P = 0.329; Fig. 2e) in this small cohort.
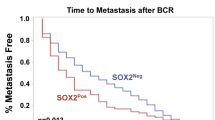
Upon further analysis of the immunohistochemistry data from the TMA samples, we observed a link between decreased level of NQO1 in carcinoma compared with normal and biochemical recurrence (BCR) following radical prostatectomy (Fig. 2f). Out of a total of 26 biochemical recurrent (BCR) patients in this TMA cohort, 23 BCR patients were subjected to cumulative NQO1 staining analysis since normal prostate tissues were not available in three cases. Of these patients, 15 (65%)/6 (26%)/2 (9%) showed a decrease/ an increase/no change respectively in NQO1 score in carcinomas. In addition, 10 (67%) out of 15 patients with low levels of NQO1 had advanced tumor (pathological stage pT3-4) while 7 (70%) of 10 in high NQO1 group had organ confined tumor (pathological stage pT2). We also used single-cell RNA sequencing expression profiles of CTCs from patients who developed BCR (GSE115501). The transcriptional scores of NQO1 is low (NQO1−) in 73% (99/136) of CTCs (Supplementary Fig. 3a). Multidimensional scaling of these patients, based on their transcriptional scores (NQO1 and TGFβ signature; Supplementary Fig. 3b), identified four groups (Fig. 2g). Groups 1 and 4 with low NQO1 expression were significantly associated with BCR (Fig. 2h; P = 2.467e-09 with Chi-square test) suggesting NQO1 attenuation in BCR patients. These findings establish the clinical significance of NQO1 in advanced prostate cancer and underscore the need to investigate the impact of reduced NQO1 expression on prostate cancer progression.
Low NQO1 is associated with mesenchymal attributes
To understand the link between NQO1 and tumor cell plasticity, we analyzed publicly available datasets of two large clinical cohorts, TCGA (n = 498) and SU2C/PCF (n = 118). Cluster heatmap of metastatic transcriptional signature with TGFβ (TGFB1, TGFBR2), Fibroblast-TGFβ response (ACTA2, COL4A1, TAGLN, SH3PXD2A) and EMT (CLDN3, CLDN7, CLDN4, CDH1, VIM, TWIST1, ZEB1, ZEB2) along with NQO1 were created. The correlation gene expression pattern showed that NQO1 expression is consistently clustered with epithelial signature and inversely correlated with TGFβ activation and mesenchymal gene signature (Fig. 3a). We then tested whether NQO1 activity is suppressed as epithelial cells undergo transition to mesenchymal phenotype. The establishment of isogenic ARCaPE (epithelial) and ARCaPM (mesenchymal) cells from parental ARCaP cells by Xu et al.32 provided an important tool to characterize crucial players involved in EMT transition. Morphologically ARCaPM cells have distinct mesenchymal characteristics including elongated appearance and dispersed cell–cell adhesion (Fig. 3b). As expected, these cells had decreased CDH1 and increased CDH2 and VIM expression (Fig. 3c) compared with ARCaPE cells. Given our previous observations that NQO1 inhibition fueled migration and androgen-independent cell survival19, we examined the involvement of NQO1 in EMT. Indeed, we found that NQO1 expression is significantly repressed in ARCaPM cells (Fig. 3c; P < 0.05). Immunoblotting and immunofluorescence analyses validated the expression observations (Supplementary Fig. 4a, b). We used cell survival against β-lapachone (β-lap) as a read-out of NQO1 functional activity since β-lap selectively kills NQO1 expressing cancer cells by generating a futile redox cycle33. ARCaPE cells were more sensitive to β-lap at both 2.5 and 5 µM doses while ARCaPM cells were significantly less sensitive with about 30% less cells killed at equivalent doses. Furthermore, the NQO1-specific inhibitor MAC220 significantly inhibited β-lap-induced cell death in ARCaPE cells (Supplementary Fig. 4c; P < 0.05 with unpaired t-test).

a Cluster heatmap of selected gene signature from two independent cohorts, TCGA and SU2C/PCF Dream Team was accessed and created using cBioPortal. b Representative microscope images of ARCaPE and ARCaPM cells showing typical morphological features associated with epithelial and mesenchymal phenotypes respectively. c Comparison of EMT marker gene expression (CDH1, CDH2, and VIM) and NQO1 between ARCaPE and ARCaPM cells by qPCR analysis (*P < 0.05). d Representative images to show morphology PC-3 cells stably transduced with non-target control (NTC) and NQO1-specific (shNQ) shRNA. e Colony formation of PC-3 NTC and shNQ cells. f Changes in epithelial (CDH1) and mesenchymal (CDH2 and VIM) markers following NQO1 KD in PC-3 cells. g Representative immunoblots of changes in EMT related markers in NTC and shNQ cells. β-actin is loading control. h Representative immunofluorescence images of changes in EMT markers in NTC and shNQ cells. All experiments were repeated thrice. Scale bars, 20 µm.
To determine whether NQO1 silencing is sufficient to induce EMT-like changes in prostate cancer cells, we used PC-3 shNQ cells because they were morphologically dispersed and more efficient in wound closure19. Indeed, NQO1 knockdown in PC-3 cells exhibited distinct morphological changes, including cell elongation and junctional disruption (Fig. 3d). PC-3 shNQ cells were slower in colony formation and the colonies formed were irregular and diffused (Fig. 3e). Consistent with these phenotypic changes, expression of CDH1 was suppressed and that of CDH2 and VIM increased (Fig. 3f). Immunoblotting and immunofluorescence showed a dramatic repression of E-cadherin, and concurrent upregulation of N-cadherin, vimentin and fibronectin protein levels in NQO1 inhibited cells (Fig. 3g, h). These observations together suggest a regulatory role for NQO1 during the transition of tumor cells from epithelial to mesenchymal phenotype.
NQO1 inhibitors increase cell migration
Analysis of cell migration by Transwell assay showed significantly increased migration in ARCaPM and PC-3 shNQ cells compared respectively with ARCaPE and PC-3 NTC cells (Fig. 4a; P < 0.05). Catalytic inhibition of NQO1 with Dicoumarol and MAC220 in PC-3 cells increased N-cadherin levels (Fig. 4b). Interrogation of the kinetics showed that MAC220 inhibited E-cadherin level for 2 days and was later restored (Fig. 4c). Restoration of E-cadherin suggests the plastic nature of these cells and may be due to the signals for constant cell rearrangement for cell–cell and matrix contact. To confirm and extend this finding, we performed cell migration assay with a catalytic NQO1 inhibitor, MAC220 that further augmented migration (p < 0.05) (Fig. 4d). Taken together, these results show that NQO1 inhibition plays a critical role in the repression of E-cadherin and enhancing the motility of prostate cancer cells.

a Representative fields of crystal violet stained images of ARCaP and PC-3 cells that migrated through 8 μm transwell plates. Scale bars, 100 µm. The bar graph shows significantly increased cell migration in NQO1low clones (ARCaPM and PC-3 shNQ) (p < 0.05). mean ± SD of n = 3 independent experiments. b Immunoblotting to show changes in EMT markers following treatment with increasing doses of NQO1 inhibitors (Dicoumarol or MAC220). β-actin was loading control. c Time kinetics of changes in E-cadherin levels following treatment of ARCaPE and PC-3 cells with MAC220 for 0–5 d. β-actin is the loading control. d Changes in migration of ARCaPM and PC-3 shNQ cells upon inhibition of NQO1 activity with MAC220 treatment (# p < 0.05). mean ± SD of n = 4 independent experiments.
NQO1 influences essential components of TGFβ signaling
Several transcription factors including SNAI1 and ZEB1 cooperate to affect E-cadherin repression. We analyzed nuclear and cytoplasmic protein fractions and found increased nuclear ZEB1 in both NQO1low-ARCaPM and PC-3 shNQ cells (Fig. 5a). Next, we investigated the possible involvement of TGFβ in PC-3 shNQ cells. We observed that the EMT-like changes observed in the morphology of PC-3 NTC cells stimulated with TGFβ1 were comparable to PC-3 shNQ cells (Supplementary Fig. 5). We found notable NQO1 inhibition by TGFβ1 stimulation (Fig. 5b). It is possible that TGFβ influences NRF2 signaling to decrease NQO1 levels34. NQO1-silencing mediated increase in N-cadherin, fibronectin, ZEB1 mimicked TGFβ1-stimulated effects on PC-3 NTC cells. Overall, NQO1 knockdown recapitulates the effect of TGFβ-activation in PC-3 cells (Fig. 5b). TGFβ-signaling was indeed activated in PC-3 shNQ cells as seen by increased expression of TGFB1 and its receptor TGFBR2. Additionally, increased expression of the latent TGFβ binding protein (LTBP, that converts the latent form to active TGFβ state) was an indication of TGFβ signaling activation (Fig. 5c). Similarly, increased expression of genes associated with active TGFβ-signaling including ZEB1, TGFB1, TGFB3, TGFBR1, and LTBP was observed in ARCaPM cells (Fig. 5d). To determine the protective role of NQO1 in EMT, we established ARCaPM cells that stably overexpress NQO1 (Supplementary Fig. 6). Expression of NQO1 partially reversed the expression of TGFβ-associated genes observed in NQO1 low ARCaPM cells (Fig. 5e). Conversely, siRNA-mediated inhibition of NQO1 in NQO1high, ARCaPE cells significantly increased TGFβ and its receptors even at 50% inhibition of NQO1 (Fig. 5f; P < 0.05). These results together suggest that NQO1 influences plasticity and migration of prostate cancer cells by restraining TGFβ signaling.

Western blotting to show changes in EMT markers in (a) cytoplasmic and nuclear extracts from ARCaP and PC-3 cells (b) whole cell lysates from PC-3 cells. c, d mRNA expression changes in TGFβ-associated genes detected by qPCR between PC-3 NTC and PC-3 shNQ cells (c) and ARCaPE and ARCaPM cells (d). e Changes in mRNA expression of TGFβ-associated gene expression following overexpression of pcDNA NQO1 (pcNQ) in ARCaPM cells. f Changes in mRNA expression of TGFβ-associated gene expression following transient knockdown of NQO1(siNQ) in ARCaPE cells. All qPCR experiments were run in duplicate and repeated three to four times.
NQO1 knockdown mimics TGFβ signaling activation
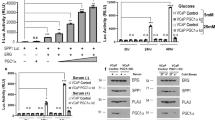
To test the hypothesis that NQO1 knockdown mimics activation of TGFβ signaling, we first determined TGFβ transcriptional activity using SBE4-Luc (four repeats of SMAD binding element; SBE). Promoter activity was significantly increased in shNQO1cells and was comparable to the effect of TGFβ1 treatment in NTC cells (P < 0.05). There was greater enhancement of luciferase activity in shNQO1 cells after TGFβ1 stimulation (Fig. 6a). LY2109761 (TGFBR1/2 inhibitor), inhibited TGFβ1-mediated SBE4-luc activation and attenuated NQO1 knockdown-mediated SBE4-luc activation in shNQO1 cells (Fig. 6a). Similarly, TGFβ transcriptional activity was also induced by NQO1 inhibition using MAC (Supplementary Fig. 7). We found the levels of SMAD3 and SMAD4 in the nucleus significantly increased in shNQO1 cells and these were further intensified in the presence of TGFβ1 (Fig. 6b and Supplementary Fig. 8; P < 0.05). TGFβ1 treatment reduced NQO1 gene expression in NTC cells while intensifying multiple TGFβ signature genes (Fig. 6c). The shNQO1 cells with higher TGFβ signature expression was inhibited by LY2109761 suggesting an interplay between NQO1 and TGFβ signaling (Fig. 6c). To characterize the extent of regulation of TGFβ signaling by NQO1, we used a focused qPCR-based array platform. Analyses of comparison between shNQO1 vs NTC and TGFβ1 vs NTC groups showed overlap of multiple TGFβ-associated genes (Fig. 6d). Additional analyses of the data using clustergram and scatter plot clearly showed that NQO1 knockdown mimics TGFβ signaling activation by regulating expression of multiple genes associated with TGFβ/BMP pathway (Supplementary Figs. 9 and 10). qRT-PCR validation with independent primers confirmed upregulation of TGFβ bonafide targets SERPINE1, SMAD7 and IGFBP3 and the downregulation of FAS. Knockdown of NQO1 and suppression of NQO1 by TGFβ1 treatment was also confirmed (Fig. 6e). As summarized in Fig. 6f, these results demonstrate that NQO1 suppresses TGFβ signaling pathway in prostate cancer cells and its suppression causes deleterious TGFβ activation perhaps by releasing the redox brake thus leading to advanced prostate cancer.

a SMAD3 and SMAD4 reporter luciferase activity in PC-3 NTC and PC-3 shNQ cells transiently transfected with SBE4-Luc containing binding sites for SMAD3 and SMAD4. mean ± SD of n = 6. p < 0.05 *, compared to PC-3 NTC; #, compared to PC-3 shNQ; $, compared to TGFβ-stimulated groups. b Immunoblotting of TGFβ response in nuclear extracts from shNTC and shNQ PC-3 cells. LaminB1 was used as a loading control for nuclear protein. All experiments were repeated a minimum of three times. c PC-3 NTC and PC-3 shNQ cells treated with TGFβ1 and LY2109761 for 48 h. Heatmap shows log2 gene expression changes detected by qPCR (n = 3). d Heatmap analysis from focused TGFβ qPCR array showing the differential gene expression between PC-3 NTC, PC-3 shNQ and PC-3 NTC + TGFβ1 treated groups (n = 2). See also Supplementary Figs. 9 and 10. e Validation of focused array by qPCR analysis with independent sets of specific primers. mean ± SD of n = 3 independent experiments. f Proposed schematic model showing how NQO1 suppresses TGFβ-induced EMT transition.
Discussion
Prostate cancer is usually diagnosed as an androgen-dependent neoplasia. However, a subset of patients frequently progress to androgen-independence and metastasis to distant sites such as the bone, which constitutes a major cause of morbidity and mortality. Therefore, it is important to understand mechanisms involved, to identify targets for effective management of advanced prostate cancer. NQO1 appeard to play contextual roles in cancer tissues. For example, it is upregulated in various types of human cancers, and in many cases, is strongly associated with tumor progression and metastasis19,35,36,37. Colorectal cancer patients with elevated NQO1 expression have been shown to correlate with high-level of HIF-1α expression and poor prognosis22. Breast and cervical cancer patients with high-level NQO1 expression are also correlated with lower disease-free survival and 5-year overall survival rates compared with those with low-level NQO1 expression21,38. While these findings indicate that NQO1 is positively associated with tumor growth and malignant progression, NQO1 is also protective against carcinogens, essential for anti-ROS defense, quinone detoxification, and metabolic stress. Compounds that induce NQO1 expression are promising strategy for cancer chemoprevention35,39. NQO1 polymorphisms, particularly C609T that causes rapid ubiquitination and degradation of the protein40, have been implicated to have higher risk of several human cancers41,42.
Given the conflicting but critical role of NQO1 that influences cancer progression, we demonstrate that NQO1 knockdown cells rapidly develop into advanced prostate adenocarcinoma. However, it remains to be validated whether NQO1 knockdown potentiates metastatic disease burden in vivo. In the future, experimental models can be used to determine if NQO1 is causal for prostate cancer cells to home to different metastatic sites given our observations of heterogeneity in downstream signaling and functional output in prostate cancer cells isolated from different metastatic sites. Further, a cohort of 150 retrospective patient samples, about half of the carcinoma samples (versus a quarter in benign/PIN groups) showed either negative or low NQO1 score. Although we did not analyze whether negative staining was associated with NQO1 polymorphism, it has been suggested that such focal loss may be due to NQO1 polymorphism and possibly associated with disease progression as indicated by concurrent PTEN loss20. Comparison of recurrence-free survival of patients based on NQO1 staining scores did not reach significance possibly due to our small cohort size. Nevertheless, assessment of publicly available expression datasets consistently demonstrated significant correlation between decreased NQO1 transcripts and poorly differentiated metastatic tumors, which was in agreement with our experimental data24,26,27,28,29,30 (Fig. 2a; Supplementary Fig. 1). Remarkably, expression data showed significant association with disease-free survival. Biochemically recurrent prostate cancer (BCR), determined by increasing prostate specific antigen (PSA) level, often occurs following initial therapy for primary cancer. Among BCR patients (n = 23) in our cohort, we noted 65% patients had decreased NQO1 levels and two thirds of them (10 out of 15) had higher stage cancer (stage 3a and b). Further, we observed more than two third of CTCs had low NQO1 which was significantly correlated with BCR outcome. It will be of interest to independently verify this observation using larger patient cohorts. Together, our results indicate that low NQO1 is associated with advanced tumor stages and high levels of NQO1 may protect from tumor progression.
In many of the cohorts in public databases, decreased NQO1 transcript levels correlated with concurrent increase in expression of mesenchymal markers such as N-cadherin, fibronectin in metastasis26,27,29,30. Therefore, we assessed the role of NQO1 in prostate cancer metastasis using prostate cancer cell culture models (PC-3 NQO1 knockdown and ARCaP EMT model systems). These cellular models along with TGFβ-stimulated PC-3 model confirmed a role for NQO1 suppression in EMT progression. Our results are particularly related with metastasis since ARCaPM is a lineage-derived from ARCaPE cells that gained 100% incidence in bone and adrenal gland metastasis in mice32. PC-3 is a bone metastasis-derived cell line and TGFβ controls multiple signaling pathways associated with bone metastasis of prostate cancer cells12. Although we did not screen many markers of EMT changes, there was no consistent change in classic EMT markers in other prostate cancer cell lines with NQO1 knockdown (Supplementary Fig. 11). Additional work is needed to determine if NQO1 plays a role in the ability of cancer cells to home to different metastatic sites given our observations of heterogeneity in downstream signaling and functional output in prostate cancer cells. However, it is fascinating to note that NQO1 expression levels are significantly low in metastatic tissues derived from multiple sites of prostate cancer patients suggesting a possible gatekeeper role against metastasis (Fig. 2a).
Using a mouse model of nonalcoholic steatohepatitis (NASH), Sharma et al.43 showed that pharmacological activation of NRF2, a master regulator of NQO1, decreases TGFβ expression. However involvement of NQO1 itself or oxidative stress is not known. Our results show that decreased NQO1 in ARCaPM clone increases TGFβ signaling and expression of mesenchymal genes, suggesting the operation of a negative feedback loop. Similarly, TGFβ signaling can be inhibited by NQO1 overexpression in the same clone. The data are consistent with increased TGFβ target genes as well as TGFβ-SMAD activation caused by NQO1 knockdown in PC-3 cells. These data provide direct evidence that NQO1 inhibits TGFβ signaling. We had reported that NQO1 silencing in hormone-responsive LNCaP cells enhanced several cytokines including IL-8 that reinforces cellular pro-migratory and pro-survival signaling19. These cytokines may potentially induce the transformation of prostate cancer cells including PC-3 cells to a more migratory/mesenchymal phenotype. Cytokine array analyses revealed that NQO1 knockdown in PC-3 cells decreased macrophage migration inhibitory factor (MIF) secretion (Supplementary Fig. 12). MIF is a pleotropic cytokine and oxidative stress sensor which modulates GSH levels by altering the cellular GSH/GSSG balance. It has recently been shown that MIF knockdown leads to the activation of the TGFβ-signaling44,45. The change in MIF further supports our notion that NQO1 silencing affects the redox balance and TGFβ signaling.
Over-treatment of low-risk prostate cancer contributed to recommendations against routine use of PSA screening. Improved risk assessment at the time of diagnosis may help reduce overtreatment by discriminating aggressive from indolent prostate cancer46. Deciphering NQO1 levels in cancer patients may be helpful to exploit the use of NQO1 targeted therapy37. On the other hand, low NQO1 levels not only show poor response to such therapies46 but also indicates disease progression (this study). Our results suggest that including NQO1 as a marker of disease progression (e.g., BCR) in molecular analyses and combining this with histopathological profiles could help stratify patients for aggressiveness of prostate cancer.
In summary, we show that NQO1 regulates TGFβ signaling to inhibit EMT and migration, which are required for prostate cancer progression. The proposed model (Fig. 6f), provides an explanation for how NQO1 overcomes the vulnerability to cellular stress, to obstruct prostate cancer cell plasticity that is essential for its progression to aggressive disease. We show that inhibition of NQO1 significantly correlated with EMT-like morphological changes and increased migration. This also increased TGFβ signaling and expression of multiple genes associated with prostate cancer progression and metastasis. NQO1 knockdown increased levels of nuclear SMADs thereby activating TGFβ-SMAD-ZEB1 signaling. Importantly, our biological data provide mechanistic details for the reported correlation between decreased NQO1 transcript and poorly differentiated metastatic prostate tumors and confirms our hypothesis that attenuation of NQO1 plays a role in TGFβ signaling mediated cancer cell plasticity.
Methods
Cell culture
Human androgen-repressed prostate cancer ARCaPE (epithelial clone) and ARCaPM (mesenchymal clone) were purchased from Novicure Biotechnology. Cells were used within eight passages of thawing. ARCaP cells were not authenticated but confirmed based on morphology, AR expression and EMT-associated genes. LNCaP and PC-3 cells were purchased from ATCC and not authenticated but were confirmed by expression or loss of AR, PSA and PTEN (Supplementary Fig. 13). ARCaP cells were cultured in MCaP-medium supplemented with 5% fetal bovine serum (FBS). LNCaP and PC-3 were cultured in RPMI1640 and F12-K, respectively. Cells were supplemented with 10% FBS, 1% antibiotics and antimycotics (GibcoBRL), and maintained in 5% CO2 at 37 °C.
Stable cell line generation
Stable NQO1 knockdown was generated as previously published19. Briefly, lentiviral particles expressing shRNA against non-targeted control (NTC) and NQO1 (shNQ) were purchased from Dharmacon (Lafayette, CO). LNCaP and PC3 cells were transduced with either NTC or shNQ in the presence of 8 μg/ml polybrene (Sigma-Aldrich) in 96-well plate. Transduction was continued for 48 h. Cells were transferred into a 24-well plate and selected with 1 μg/ml puromycin (Sigma-Aldrich). Serially diluted cells were grown and efficiency of transduction was confirmed in isolated clones by detecting GFP fluorescence. For transient knockdown, cells were transfected with either scramble or NQO1 SMARTpool siRNA (Dharmacon) using Lipofectamine (Invitrogen). Knockdown was verified by qPCR and immunoblot analysis. For stable overexpression, cells were transfected with pcDNA3.1 vector control or pcDNA-NQO1 (pcNQ). After selection with G418, NQO1 overexpression was verified by western blotting.
Animal experiments
For orthotopic implantation of human prostate cancer cells, male athymic nude mice (4–6 weeks of age) were obtained from Charles River Laboratories. After one-week acclimation period, mice were randomized into two groups (n = 8 per group) and implanted orthotopically with 1 × 106 LNCaP cells either expressing stably silenced NQO1 (LNCaP shNQ) or non-targeted control (LNCaP NTC) in matrigel (1:1 v/v, Corning). Tumor development was examined by palpation at the site of injection weekly and body weight was measured weekly. The experiment was terminated when the first palpable tumors were noted irrespective of group. Mice were euthanized and all tissues including serum were collected. One animal in the shNQ group died from non-tumor related cause. The prostates were observed for gross tumor, weighed and photographed. A portion of each prostate tissue was snap-frozen and the remaining formalin-fixed and paraffin-embedded. Following Hematoxylin and Eosin staining of the embedded tissue, histopathological evaluation was conducted by a blinded pathologist. All animal procedures were approved by UTHSCSA IACUC.
Bioinformatics data analysis
Microarray dataset of NQO1 knockdown and nontargeted control LNCaP cells (GSE58336) were analyzed by DAVID web tool. The Cancer Genome Atlas (TCGA) and other publicly available datasets were accessed. Expression of NQO1 in primary and metastatic patient samples was queried using Oncomine database (Chandran24, Yu26, Grasso25, Lapointe27, Ramaswamy28, Ramaswamy229, and Tamura30 prostate studies available through Oncomine http://www.oncomine.org/). Kaplan–Meier analysis was extracted (GSE40272; GSE70769) and analyzed from PROGgeneV2. Cluster heatmap data were accessed and analyzed using cBioPortal for TCGA and SU2C/PCF Dream Team.
Prostate tumor specimens and immunohistochemical analysis
A set of TMA consisting of 150 radical prostatectomy specimens was constructed as described previously at the University of Rochester Medical Center47. Appropriate approval was obtained from the URMC institutional review board for the construction and use of the TMA. The tissue sections were immunohistochemically stained with anti-NQO1 along with relevant controls as described previously19. Immunoreactive scores were calculated as negative (0) weakly positive (1+), moderately positive (2+), and strongly positive (3+), as described earlier47, via considering both staining intensity and the percentage of Immunoreactive cells. Recurrence-free survival was compared between high (2/3+) and low NQO1 (0/1+) expression groups.
Circulating tumor cells (CTCs)
Subjects were enrolled in a longitudinal, observational clinical study called Longitudinal Study of Prostate Cancer Determinants of Resistance (Los Padres). After obtaining UTHSCSA IRB consent, we obtained blood (8-16 mL per patient) for CTC isolation with the following modification48. The peripheral blood mononuclear cell (PBMC) interphase portion was collected following Ficoll cushion centrifugation. MojoSort™ Human CD45 Nanobeads (BioLegend, #480030) was used to deplete CD45+ cells. To enrich CTCs, we cultured cells in PRIME-XV tumorsphere medium (Irvine Scientific, #91130) and then expanded into tumor spheres for 1 to 2 weeks. A nanobead depletion assay was carried out to remove CD45-positive cells before cDNA generation.
Single-cell transcriptomic sequencing, mapping, and clustering of CTCs
Viable cells (detected with LIVE/DEAD Viability/Cytotoxicity Kit; Invitrogen) were loaded in Fluidigm C1 single-cell mRNA seq IFC (Fluidgm) for single-cell isolation. Single-cell cDNA synthesis was carried out with SMART-Seq v4 Ultra Low Input RNA Kit (Clontech). Single-cell cDNA synthesized from each living cell was used in RNA-seq library preparation per the manufacturer’s protocol. Nextera XT DNA Library Prep Kit (Illumina) with Nextera XT Index Kit v2 Set A and B index primers were used for cDNA tagmentation using diluted cDNA (0.3 ng/μL). Paired-end cDNA sequencing was carried out on the HiSeq 3000 system (Illumina). Base calling was performed using the Illumina Real-Time Analysis software. Output paired-end FASTQ files were aligned to the GRCh38 reference genome using STAR alignment software49. Gene expression levels were quantified by applying RSEM50, and fragments per kilobase of transcript per million (FPKM) mapped reads were calculated. Cells with four leucocyte markers (displaying low uniquely aligned reads and high expression; PTPRC, CD34, CD19, and FCGR3A) were excluded from further analysis. t-Distributed Stochastic Neighbor Embedding (t-SNE) (R package Rtsne) was used to generate scatter plot of 136 CTCs based on NQO1 and TGFβ transcriptomic profile (log FPKM + 1), and mclust algorithm was subsequently applied to robustly differentiate CTCs into different clusters.
Cell viability, colony formation, and migration assays
Cell viability and colony formation assays were performed as described previously19. Briefly, the MTT assay is used to determine the cell viability. Absorbance was measured and normalized to express cell viability in percent of control. To assess colony formation 1 × 103–5 × 103 cells were seeded in triplicate in 6-well plate. After 9–12 days, colonies were fixed, stained with 0.05% crystal violet in methanol. Plates were scanned to obtain digital images. To determine chemotactic migration through a 8 µm pore size insert (Corning), 50 µL cell suspension (5 × 104 cells in serum free medium) was dispensed in the upper insert and allowed to migrate towards 10% serum containing medium on the bottom well.
Cytokine array
Antibody-based array was used to determine cytokines in culture supernatant according to the manufacturer's protocol (R&D Systems).
RNA extraction and real-time quantitative PCR (qPCR)
Total RNA was extracted and used to transcribe into cDNA as published21. Cells were harvested and total RNA was extracted using TRIzol according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). Isolated RNA was reverse transcribed into cDNA using SuperScript VILO cDNA Synthesis Kit (Invitrogen). Primer pairs (Supplementary Table 4) were purchased from Integrated DNA Technologies. Gene expression levels were analyzed by qPCR on CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories, CA). A standard program: 95 °C for 10 min, 40 cycles at 95 °C for 15 s and 60 °C for 1 min was used for all qPCR assays. An additional melting step was added to analyze the specificity of the obtained amplification products. Each Ct value was normalized to β-actin levels as reference.
Transient transfection and luciferase activity
Stable clones of PC-3 NTC and PC-3 shNQ were co-transfected with SBE4-Luc51 (Addgene) and pRL-TK (Promega) using Lipofectamine 2000 according to the manufacturer’s instruction (Invitrogen). Cells were plated at a density of 1 × 105 cells/well in 24-well plate. After 24 h, cells were transfected with 0.5 µg of promoter-linked luciferase vector and 10 ng of pRL-TK vector in OptiMEM medium (Invitrogen). Luciferase activity was determined using the dual luciferase reporter assay (Promega, Madison, WI), and emitted light was measured with a luminometer (Turner Biosystems, Sunnyvale, CA).
qPCR array
TGFβ gene expression was analyzed using qPCR arrays (Human TGFβ/BMP Signaling Pathway RT2 Profiler™ PCR Array, QIAGEN, Cat. # PAHS-035Y) on CFX96 Real-Time PCR Detection System per manufacturer’s instructions.
Western blot analysis
Whole cell extracts were prepared using 2 × SDS-containing-Laemmli buffer. Cytoplasmic and nuclear extracts were prepared using the NE-PER nuclear extraction kit (Pierce). Equal amounts of total, cytoplasmic or nuclear extracts were resolved by SDS-PAGE and immunoblotted with specific primary antibodies. Primary antibodies were diluted in 5% non-fat milk or 3% bovine serum albumin (BSA) in Tris-buffered saline with 0.1% Tween-20 (TBS-T) at the following dilutions: 1:500 (PTEN, HDAC1, Fibronectin); 1:1000 (AR, α-tubulin, TCF8/ZEB1, N-cadherin); 1:2000 (PSA, E-cadherin, Vimentin, β-catenin, Smad2, Smad3, Smad2/3, Smad4); 1:7500 (NQO1, β-actin, LaminB1). Proteins were visualized using ECL kit (Perkin Elmer) and digitally processed using Syngene G-Box (Syngene, Frederick, MD). Membranes were stripped and re-probed with β-actin and Lamin B1 antibodies for total and nuclear loading controls respectively.
Immunofluorescence
Cells were grown on cell culture-treated coverglass and immunostained with specific antibodies at the following dilution: 1:100 (N-cadherin); 1:250 (E-cadherin, Vimentin) and 1:500 (NQO1). Immunofluorescence was performed as described previously19. Immunostained cells were examined on a Sweptfield confocal system (Prairie Technologies, Middleton, WI) equipped with a Nikon Ti microscope.
Statistics and reproducibility
Measurements were made on distinct samples. Difference between groups was analyzed by ANOVA or Student’s t-test using GraphPad Prism software. TGFβ qPCR array was performed twice. For all other in vitro assays, three independent experiments were performed to confirm reproducibility. In clinical samples and public data sets, sample size was limited by availability. In in vivo mouse and in vitro assays, sample sizes were chosen based on our experience and previously published literature. Fisher's exact test was used to analyze significance of the associations between disease grade, clinicopathological features and NQO1 staining levels. For survival analysis, Kaplan–Meier curves were generated using either PROGgeneV2 or Prism software and log-rank analysis was performed. Results are expressed as mean ± SD. P values < 0.05 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Our microarray data set of NQO1 knockdown and non-targeted control LNCaP cells (GSE58336) and RNA sequencing data from CTCs (GSE115501) are available in Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/). NQO1 expression in patient samples from multiple cohorts referenced in the study are available in Oncomine (http://www.oncomine.org/). The TCGA and SU2C/PCF data are publicly available (http://www.cbioportal.org/). Kaplan–Meier analysis of two cohorts (GSE40272; GSE70769) are available in PROGgeneV2 (http://www.compbio.iupui.edu/proggene). All other data supporting the findings of this study are available within the article and its supplementary information files. Raw data are provided as Supplementary Data 1.
References
Ross, D. et al. NAD(P)H:quinone oxidoreductase 1 (NQO1): chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem. Biol. Interact. 129, 77–97 (2000).
Asher, G., Lotem, J., Kama, R., Sachs, L. & Shaul, Y. NQO1 stabilizes p53 through a distinct pathway. Proc. Natl Acad. Sci. USA 99, 3099–3104 (2002).
Dinkova-Kostova, A. T. & Talalay, P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys. 501, 116–123 (2010).
Izumi, K. et al. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO Mol. Med. 5, 1383–1401 (2013).
Lamouille, S., Xu, J. & Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196 (2014).
Marin-Aguilera, M. et al. Epithelial-to-mesenchymal transition mediates docetaxel resistance and high risk of relapse in prostate cancer. Mol. Cancer Ther. 13, 1270–1284 (2014).
Li, L. & Li, W. Epithelial-mesenchymal transition in human cancer: comprehensive reprogramming of metabolism, epigenetics, and differentiation. Pharm. Ther. 150, 33–46 (2015).
Derynck, R., Akhurst, R. J. & Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 29, 117–129 (2001).
Padua, D. & Massague, J. Roles of TGFbeta in metastasis. Cell Res. 19, 89–102 (2009).
Karantanos, T., Corn, P. G. & Thompson, T. C. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 32, 5501–5511 (2013).
Pickup, M., Novitskiy, S. & Moses, H. L. The roles of TGFbeta in the tumour microenvironment. Nat. Rev. Cancer 13, 788–799 (2013).
Fournier, P. G. et al. The TGF-beta signaling regulator PMEPA1 suppresses prostate cancer metastases to bone. Cancer Cell 27, 809–821 (2015).
Mariathasan, S. et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018).
Tauriello, D. V. F. et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554, 538–543 (2018).
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 68, 7–30 (2018).
Thapa, D. & Ghosh, R. Antioxidants for prostate cancer chemoprevention: challenges and opportunities. Biochem. Pharm. 83, 1319–1330 (2012).
Giannoni, E., Parri, M. & Chiarugi, P. EMT and oxidative stress: a bidirectional interplay affecting tumor malignancy. Antioxid. Redox Signal 16, 1248–1263 (2012).
Kumar, B., Koul, S., Khandrika, L., Meacham, R. B. & Koul, H. K. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 68, 1777–1785 (2008).
Thapa, D. et al. NQO1 suppresses NF-kappaB-p300 interaction to regulate inflammatory mediators associated with prostate tumorigenesis. Cancer Res. 74, 5644–5655 (2014).
Kurfurstova, D. et al. DNA damage signalling barrier, oxidative stress and treatment-relevant DNA repair factor alterations during progression of human prostate cancer. Mol. Oncol. 10, 879–894 (2016).
Yang, Y. et al. Clinical implications of high NQO1 expression in breast cancers. J. Exp. Clin. Cancer Res. 33, 14 (2014).
Oh, E. T. et al. NQO1 inhibits proteasome-mediated degradation of HIF-1alpha. Nat. Commun. 7, 13593 (2016).
Thapa, D. & Ghosh, R. Chronic inflammatory mediators enhance prostate cancer development and progression. Biochem Pharm. 94, 53–62 (2015).
Chandran, U. R. et al. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer 7, 64 (2007).
Grasso, C. S. et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 487, 239–243 (2012).
Yu, Y. P. et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J. Clin. Oncol. 22, 2790–2799 (2004).
Lapointe, J. et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl Acad. Sci. USA 101, 811–816 (2004).
Ramaswamy, S. et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc. Natl Acad. Sci. USA 98, 15149–15154 (2001).
Ramaswamy, S., Ross, K. N., Lander, E. S. & Golub, T. R. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 33, 49–54 (2003).
Tamura, K. et al. Molecular features of hormone-refractory prostate cancer cells by genome-wide gene expression profiles. Cancer Res. 67, 5117–5125 (2007).
Goswami, C. P. & Nakshatri, H. PROGgeneV2: enhancements on the existing database. BMC Cancer 14, 970 (2014).
Xu, J. et al. Prostate cancer metastasis: role of the host microenvironment in promoting epithelial to mesenchymal transition and increased bone and adrenal gland metastasis. Prostate 66, 1664–1673 (2006).
Dong, Y. et al. Prostate cancer radiosensitization through poly(ADP-Ribose) polymerase-1 hyperactivation. Cancer Res. 70, 8088–8096 (2010).
Ryoo, I. G., Shin, D. H., Kang, K. S. & Kwak, M. K. Involvement of Nrf2-GSH signaling in TGFbeta1-stimulated epithelial-to-mesenchymal transition changes in rat renal tubular cells. Arch. Pharm. Res. 38, 272–281 (2015).
Oh, E. T. & Park, H. J. Implications of NQO1 in cancer therapy. BMB Rep. 48, 609–617 (2015).
Li, Z. et al. NQO1 protein expression predicts poor prognosis of non-small cell lung cancers. BMC Cancer 15, 207 (2015).
Huang, X. et al. Leveraging an NQO1 bioactivatable drug for tumor-selective use of poly(ADP-ribose) polymerase inhibitors. Cancer Cell 30, 940–952 (2016).
Ma, Y. et al. NQO1 overexpression is associated with poor prognosis in squamous cell carcinoma of the uterine cervix. BMC Cancer 14, 414 (2014).
Bauman, J. E. et al. Prevention of carcinogen-induced oral cancer by sulforaphane. Cancer Prev. Res (Philos.) 9, 547–557 (2016).
Siegel, D. et al. Rapid polyubiquitination and proteasomal degradation of a mutant form of NAD(P)H:quinone oxidoreductase 1. Mol. Pharm. 59, 263–268 (2001).
Ross, D. & Siegel, D. NAD(P)H:quinone oxidoreductase 1 (NQO1, DT-diaphorase), functions and pharmacogenetics. Methods Enzymol. 382, 115–144 (2004).
Chao, C., Zhang, Z. F., Berthiller, J., Boffetta, P. & Hashibe, M. NAD(P)H: quinone oxidoreductase 1 (NQO1) Pro187Ser polymorphism and the risk of lung, bladder, and colorectal cancers: a meta-analysis. Cancer Epidemiol. Biomark. Prev. 15, 979–987 (2006).
Sharma, R. S. et al. Experimental nonalcoholic steatohepatitis and liver fibrosis are ameliorated by pharmacologic activation of Nrf2 (NF-E2 p45-related factor 2). Cell. Mol. Gastroenterol. Hepatol. 5, 367–398 (2018).
Liu, L. et al. A global genomic view of MIF knockdown-mediated cell cycle arrest. Cell Cycle 7, 1678–1692 (2008).
Heinrichs, D. et al. Macrophage migration inhibitory factor (MIF) exerts antifibrotic effects in experimental liver fibrosis via CD74. Proc. Natl Acad. Sci. USA 108, 17444–17449 (2011).
Gerber, D. E. et al. Phase 1 study of ARQ 761, a beta-lapachone analogue that promotes NQO1-mediated programmed cancer cell necrosis. Br. J. cancer 119, 928–936 (2018).
Izumi, K. et al. Seminal plasma proteins in prostatic carcinoma: increased nuclear semenogelin I expression is a predictor of biochemical recurrence after radical prostatectomy. Hum. Pathol. 43, 1991–2000 (2012).
Drost, J. et al. Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc. 11, 347–358 (2016).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 12, 323 (2011).
Zawel, L. et al. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol. cell 1, 611–617 (1998).
Acknowledgements
This work was supported by 5R01CA149516 (R.G.), 1R01AT7448 and CTRC 40th Anniversary Distinguished Professor of Oncology Endowment (A.P.K.). D.T. was supported in part by an American Cancer Society (Joe and Jessie Crump Foundation Medical Research Fund) Postdoctoral Fellowship (PF-15-219-01-CSM). S.B.H. was supported in part by a predoctoral fellowship from the Cancer Prevention Research Institute of Texas (RP170345). C.L.C. was supported in part by the Cancer Prevention Research Institute of Texas, RP150600 (T.H.-M.H.), 5U54CA113001-10-10 (T.H.-M.H.), DoD PC170821 (T.H.-M.H.) and the V-Foundation Translational Award. We thank the Bioanalytic Single-Cell Core (CPRIT) at UTHSCSA for partial support of data analysis. We thank Dr. David Ross (University of Colorado Medical School, Denver, CO) for NQO1 reporter and MAC220. We also thank the Optical Imaging shared resource at UTHSCSA.
Author information
Authors and Affiliations
Contributions
All authors of this paper have read and approved the final version of the submitted manuscript. D.T., A.P.K., and R.G. developed the original hypothesis, conceived the study, and designed experiments. D.T. performed most of the in vitro experiments and analyzed data. S.-B.H., A.R.M., and X.Y. performed the animal experiments and analyzed data. R.G.B. performed immunohistochemistry. C.-N.H., C.-.L.C., and T.H.-M.H. analyzed RNA-seq data from circulating tumor cells. M.A.L. and H.M. provided clinical specimens, and H.M. also analyzed and interpreted data. R.L.R. performed pathological analysis on mouse tissues. D.T. and R.G. wrote and edited the manuscript. R.G. supervised the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thapa, D., Huang, SB., Muñoz, A.R. et al. Attenuation of NAD[P]H:quinone oxidoreductase 1 aggravates prostate cancer and tumor cell plasticity through enhanced TGFβ signaling. Commun Biol 3, 12 (2020). https://doi.org/10.1038/s42003-019-0720-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-019-0720-z
This article is cited by
-
Anoikis patterns via machine learning strategy and experimental verification exhibit distinct prognostic and immune landscapes in melanoma
Clinical and Translational Oncology (2023)
-
Review on NAD(P)H dehydrogenase quinone 1 (NQO1) pathway
Molecular Biology Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
