
Abstract
Background:
Poly adenosine diphosphate (ADP)-ribose polymerase (PARP) is essential in cellular processing of DNA damage via the base excision repair pathway (BER). The PARP inhibition can be directly cytotoxic to tumour cells and augments the anti-tumour effects of DNA-damaging agents. This study evaluated the optimally tolerated dose of olaparib (4-(3--4-fluorophenyl) methyl-1(2H)-one; AZD2281, KU0059436), a potent PARP inhibitor, with dacarbazine and assessed safety, toxicity, clinical pharmacokinetics and efficacy of combination treatment.
Patients and methods:
Patients with advanced cancer received olaparib (20–200 mg PO) on days 1–7 with dacarbazine (600–800 mg m−2 IV) on day 1 (cycle 2, day 2) of a 21-day cycle. An expansion cohort of chemonaive melanoma patients was treated at an optimally tolerated dose. The BER enzyme, methylpurine-DNA glycosylase and its substrate 7-methylguanine were quantified in peripheral blood mononuclear cells.
Results:
The optimal combination to proceed to phase II was defined as 100 mg bd olaparib with 600 mg m−2 dacarbazine. Dose-limiting toxicities were neutropaenia and thrombocytopaenia. There were two partial responses, both in patients with melanoma.
Conclusion:
This study defined a tolerable dose of olaparib in combination with dacarbazine, but there were no responses in chemonaive melanoma patients, demonstrating no clinical advantage over single-agent dacarbazine at these doses.
Similar content being viewed by others


Main
One of the primary roles of poly adenosine diphosphate (ADP)-ribose polymerase-1 (PARP-1) is to detect DNA strand breaks that occur following DNA damage. Activation of PARP-1 in this setting results in DNA repair via the base excision repair pathway (BER) (Virag and Szabo, 2002). The PARP-1 activity is frequently increased in tumour cells, with evidence that inhibition of PARP can be cytotoxic (Chalmers, 2009). The PARP inhibition leads to accumulation of DNA single-strand breaks resulting in DNA double-strand breaks at replication forks. The PARP-1 inhibition augments the anti-tumour effects of many DNA-damaging cytotoxic agents or radiation (Chalmers, 2009).
Olaparib (4-(3--4-fluorophenyl) methyl-1(2H)-one; AZD2281) is a potent, orally active, PARP inhibitor (Menear et al, 2008). Olaparib combined with the methylating agent temozolomide, a pro-drug of 3-methyl-(triazen-1-yl-4-carboxamide (MTIC), enhances cytotoxicity in tumour cell lines and xenografts (Menear et al, 2008). Dacarbazine, dimethyltriazenoimidazole carboxamide (DTIC) which is also activated to form MTIC, was selected for clinical investigation in combination with olaparib to explore the combination's activity in melanoma.
DNA adducts, such as 3-methyladenine (3meA) and N7-methylguanine (7-meG), that are produced in DNA following exposure to dacarbazine are processed by the BER pathway (Fromme et al, 2004). A damage-specific DNA glycosylase removes the alkylated DNA base, resulting in the generation of apurinic sites that are cleaved by the abundant endonuclease, APE, and the resulting single-strand breaks are repaired by the coordinated intervention of PARP, DNA polymerase β, X-ray repair cross-complementing-1 and ligases I and III (Dantzer et al, 1999). The PARP senses and is activated by the DNA single strand breaks and catalyses poly ADP ribosylation and consequent activation of the various proteins involved in the BER pathway.
The aim of combining dacarbazine with olaparib was to disrupt BER function, and thus elicit an accumulation of strand breaks and ultimately increased cytotoxicity. Given the requirement of single-strand break formation in this process, PARP inhibition may have a greater impact in cells with a greater ability to remove the damaged bases, that is, with higher N-methylpurine-DNA glycosylase (MPG) activity. This activity may itself be upregulated by combination treatment with a PARP inhibitor and an alkylating agent, and this may have consequences for cell death, as 3meA is a toxic DNA lesion (Tentori et al, 1999, 2001).
The rationale for this study (KU36-73, NCT00516802) was that intermittent dosing of olaparib, given in combination with dacarbazine, would sufficiently inhibit the PARP-1 repair of methylated DNA to produce an enhanced clinical effect. Primary objectives were to determine the safety, tolerability and dose-limiting toxicity (DLT) of a combination of oral olaparib and intravenous dacarbazine in patients with advanced solid tumours. Secondary objectives were to determine the pharmacokinetic profile of olaparib in combination with dacarbazine, to investigate the pharmacokinetic–pharmacodynamic profile of the combination in surrogate tissues and to enable a preliminary assessment of their anti-tumour activity. Thus both inter- and intraindividual variations in MPG activity during the course of the KU-DTIC treatment cycle were measured. Levels of 7-meG in DNA were quantified before treatment and following DTIC as both an indicator of the level of alkylation damage generated and an indicator of the amounts lost through repair or cell turnover, respectively.
Patients and methods
The study was conducted in accordance with the Principles of the International Conference on Harmonization of Good Clinical Practice guidelines and the Declaration of Helsinki. The protocol was approved by an independent ethics committee, according to UK and local requirements. All patients enroled in the study gave informed written consent (KU36-73, NCT00516802).
Individuals aged ⩾18 years with a life expectancy of at least 3 months were eligible for the study. The ECOG performance status of 2 or better, adequate hepatic, renal and bone marrow function, and platelet count were required. For the initial dose-escalation cohorts, patients had to have a histologically or cytologically confirmed malignant solid tumour refractory to standard therapy. At a dacarbazine dose of 800 mg m−2 in the escalation phase and in the dose-expansion phase, only patients with unresectable stage III/IV cutaneous or unknown primary melanoma and no previous systemic cytotoxic chemotherapy were allowed to participate.
Treatment
Olaparib (AstraZeneca, Macclesfield, UK) was administered orally as capsules twice daily on days 1–7 of each 21-day treatment cycle. In cycle 2, olaparib was administered on days 2–8 to permit the pharmacokinetics of dacarbazine given alone to be determined. Dacarbazine (Bayer, Bedford, UK) was administered by intravenous infusion over 60 min on day 1 of each cycle, 3 h after olaparib administration (except in cycle 2).
Study design
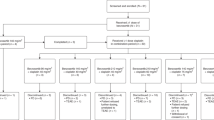
The starting dose was olaparib 10 mg bd with 600 mg m−2 dacarbazine (10: 600: combination doses will be abbreviated in this way for the remainder of the manuscript). The dose for successive cohorts was modified according to the scheme presented in Figure 1 taking into account the toxicities experienced by preceding patient cohorts.

Chronological flow chart of implemented study design cohorts.
The DLT was defined as any of the following: grade 4 haematological toxicity lasting ⩾5 days; grade 3 or 4 febrile neutropaenia; and grade 3 or 4 non-haematological toxicity. However, grade 3 or greater non-haematological toxicities were not classified as DLTs, if the nature and severity of the toxicity was attributable to DTIC alone. If one out of three patients at a dose level developed DLT, up to three additional patients were treated at that dose level. If one out of the additional patients developed DLT, dose escalation ceased and a preceding dose level or an intermediate dose level was tested. This lower dose was defined as the maximum tolerated dose for that dose of dacarbazine, unless ⩾2 out of 6 patients developed DLT.
The dose-expansion phase involved 10 patients treated at the selected olaparib and dacarbazine doses, with the option to move to a randomised expansion phase, if a 20% overall response rate was observed.
Toxicity and response evaluation
Safety assessments included physical examination, chemistry, haematology and urinalysis. Toxicities were evaluated at least weekly during the study period and were graded according to the National Cancer Institute Common Toxicity Criteria version 3 (Tsuchida and Therasse, 2001). Efficacy was assessed every other cycle.
Pharmacodynamics
The MPG activity and levels of 7-meG in DNA were determined in peripheral blood mononuclear cells before and following treatment with dacarbazine. Samples were obtained on days 1 and 8 of the treatment cycle, and analysed according to previously published methods. The MPG activity in the cell extract was quantified by an oligonucleotide cleavage assay. The amounts of 7-meG in peripheral blood DNA were quantified according to an immunoblot method (Harrison et al, 2001).
N-methylpurine-DNA glycosylase activity
Whole blood (1 ml) was thawed, centrifuged at 3700 g for 10 min and the supernatant discarded. The cell pellet was resuspended in buffer, and extracts prepared by sonication as described by Harrison et al (2001). The MPG activity in the cell extract was quantified by an oligonucleotide cleavage assay. Briefly, an oligonucleotide containing ethenoadenine (a substrate for MPG) close to the 5′-end was labelled with 32P γATP, annealed to its 5′-biotinylated complement and immobilised on a streptavidin-coated 96-well plate. Incrementally increasing amounts of cell extract based on DNA content as quantified by picogreen-based assay (Invitrogen Ltd., Paisley, UK) were incubated with the substrate oligonucleotide for 3 h at 37°C. The 32P-labelled 5-mer released into the supernatant as a result of removal of ethenoadenine by MPG, and the subsequent action of APE were quantified on a TOP COUNT machine (Perkin Elmer LAS, Beaconsfield, UK). Specific activity was calculated as femtomoles (Fmoles) ethenoadenine removed per μg of extract DNA per hour. The lower limit of quantitation (LLOQ) was defined as 2.3 Fmoles ethenoadenine removed per μg DNA per hour.
N7-methylguanine in DNA
The amounts of 7-meG in peripheral blood DNA were quantified in pre- and post-DTIC treatment (day 1 at 5 h and day 8) samples according to the immunoblot method previously described (Menear et al, 2008). The chemiluminescence signal was generated by incubation with ECL-Advance (Amersham, Chalfont St Giles, UK), detected with the Chemi Genius Bio Imaging System (Syngene, Cambridge, UK) and quantified using Genetools software (Syngene). The LLOQ was defined as 0.3 Fmoles 7-meG per μg DNA.
Pharmacokinetics
Venous blood was drawn for determination of pharmacokinetic profiles for olaparib and dacarbazine when dosed alone and in combination. Blood samples were collected before the patients started taking olaparib and 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8 and 9–10 h after taking the drug on day 1 of cycle 1. Blood samples were also collected before the start of the dacarbazine infusion and 0.5, 1, 2 and 5–6 h after the end of infusion to quantify plasma levels of dacarbazine on day 1 of cycles 1 and 2. Plasma dacarbazine and olaparib concentrations were determined by high-performance liquid chromatography with tandem mass spectrometric detection.
Results
A total of 40 patients were enroled in the study at three centres, and their characteristics are summarised in Tables 1 and 2. All 40 patients were evaluable for toxicity and for tumour response.
Dose escalation and extent of exposure
A total of 153 cycles (median 2.5 cycles per patient) of olaparib and dacarbazine were administered in the study. There were no dose reductions of olaparib, although six patients were non-compliant and missed one dose of their olaparib treatment each. Four patients required dacarbazine dose reductions of 200 mg m−2 and in one patient, a second dose reduction was needed because of haematological toxicity.
During the dose-escalation phase of the trial, three cohorts of patients were successfully treated without major potentiation of toxicity or serious side effects. When patients were treated with 40: 800 in part II, a higher incidence of bone marrow toxicity, particularly neutropaenia, was noted compared with rates reported for single-agent DTIC. Although the toxicities were not dose limiting per protocol, it was apparent that the dose combination of 40: 800 could not be sustained over several cycles. Therefore, the protocol was amended to allow a dose combination of 600 mg m−2 DTIC in combination with increasing olaparib doses from 40 to 200 mg bd to be explored.
Three patients in the dose-escalation phase experienced a DLT during the first treatment cycle. One occurred in cohort 4 (40: 800) and two in cohort 6 (200: 600). The highest combination doses that were deliverable were 100: 600 and 20: 800. Maximal PARP inhibition was observed at doses above 60 mg bd in single-agent trials so the 100: 600 dose combination was chosen for the dose-expansion component of the study (Fong et al, 2009).
Safety
Adverse events were as anticipated for dacarbazine (Table 3). Most frequent toxicities were neutropaenia and anaemia, although nausea, fatigue, anorexia, diarrhoea and thrombocytopaenia were common. As anticipated, the incidence of neutropaenia was higher than that observed with single-agent dacarbazine, affecting 10 patients and in 9 of these, the severity was ⩾grade 3. Two patients died within 30 days of receiving study drug. Neither death was considered to be related to the combination treatment: one because of myocardial ischaemia and the other to pneumonia in the setting of progressive disease.
Dose-limiting toxicities
One patient treated with 40: 800 experienced grade 3 hypophosphataemia and grade 3 leucopoenia. Cycle 2 was delayed because of thrombocytopaenia, which peaked at grade 3 and did not recover until day 49. The patient was taken off study because of these toxicities. Two patients treated with 200: 600 experienced grade 4 neutropaenia on day 15, recovering by day 22. This was considered to be dose limiting, although the duration of neutropaenia could not be confirmed. These two dose levels were not chosen for further evaluation because of the toxicities observed in these cohorts, in particular the occurrence of neutropaenia in one-third of patients in each of the cohorts with insufficient recovery before the date of the next planned dacarbazine infusion.
Efficacy
All patients were evaluable for efficacy. Two partial responses were documented, and eight patients had stable disease where follow-up measurements must have met the stable disease criteria at least once after trial entry at a minimum interval of 12 weeks. The other 30 patients progressed on treatment (Table 4). Both partial responses were seen in patients with previously treated melanoma: one where the patient had received five previous lines of therapy and the other where they had received one previous regimen.
The median time to disease progression was 82 days (95% confidence interval (CI): 38–108 days) in the dose-escalation phase in refractory solid tumour patients and 42 days (95% CI: 36–84 days) for chemotherapy-naive melanoma patients. The median time to disease progression overall was 43 days (95% CI: 36–108 days).
Pharmacokinetics
A total of 29 patients provided evaluable pharmacokinetic data for olaparib with or without dacarbazine. The co-administration of dacarbazine appeared to have had little or no effect on exposure to olaparib, with respect to peak concentration (Cmax) or area under the concentration–time curve. Similarly, in 17 evaluable patients, there was no discernable impact of olaparib on dacarbazine pharmacokinetics.
Pharmacodynamics
Blood samples collected from 24 patients were available for analysis. For individual patients, MPG activities did not vary significantly or consistently over the course of the sampling (Figure 2A), indicating that MPG activity was not substantially affected by the doses and schedules of olaparib and dacarbazine used in this study, or sampling times. Mean MPG activity, based on all times points measured, showed marked interpatient variation, with values ranging from 3.3 to 16.2 Fmol per μg DNA per hour (mean±s.d. 8.7±6.0; Figure 2B).

Pharmacokinetic data: (A) Activity of N-methylpurine-DNA glycosylase (MPG) during course of sampling after olaparib administration. (B) Mean MPG activity for 21 available patients. (C) Effect of MPG activity on 7-methylguanine (7-meG) levels.
Levels of 7-meG before treatment were less than the LLOQ for 14 out of the 20 patients analysed, with very low levels in the other 6 individuals (range 0.3–1.6 Fmol per μg DNA). Mean 7-meG levels rose significantly (P<0.01) on day 1, 5 h after dacarbazine administration (50.5±8.2 Fmol per μg DNA, n=24), and remained above baseline at day 8 (14.6±5.4 Fmol per μg DNA, n=5). This is consistent with previous observations made following temozolomide treatment (Watson et al, 2009) that levels reduce over time, most likely because of cell turnover and/or removal of 7-meG by MPG. Mean 7-meG levels after treatment were higher in patients treated with 800 mg m−2 dacarbazine (55.3±8.1 Fmol per μg DNA) than those treated with 600 mg m−2 (47.4±8.3 Fmol per μg DNA) (P<0.05). There was no correlation between MPG activity and 7-meG levels after dacarbazine administration in individual patients (Figure 2C). Unfortunately, no data were available from the two patients who had partial responses on treatment.
Discussion
Treatment options for patients with metastatic melanoma are limited, with existing therapies only producing low rates of objective radiological responses and short periods of clinical benefit. Dacarbazine remains the standard of care and is the benchmark against which other therapies are compared.
This is the first study evaluating the optimally tolerated dose of olaparib, a potent PARP inhibitor, in combination with dacarbazine in patients with either advanced solid tumours or chemotherapy-naive melanomas. Adverse event rates were similar to those previously reported for dacarbazine alone at doses from 800 to 1000 mg m−2, with the exception of neutropaenia. Myelosuppression occurs in about 25% of patients treated with dacarbazine, but grade 3 or 4 events are seen in only 1–2% (Bajetta et al, 1994; Middleton et al, 2000). As expected from other studies with PARP inhibitors in combination with temozolomide and pre-clinical studies with olaparib significant myelotoxicity, especially neutropaenia was more frequent and defined the dose of the two drugs for combined use. Grade 3 or 4 neutropaenia affected nine patients (22.5%) in this study, and appeared more frequent with higher doses of dacarbazine or olaparib. The increased neutropaenia, although readily managed, led to an increase in delays in the administration of the second cycle of therapy and reductions in the dose of dacarbazine in the highest dose cohorts.
The 100 mg bd olaparib and 600 mg m−2 dacarbazine dose combination was chosen for the dose confirmation component of the study. Monotherapy olaparib studies had indicated that doses of 100 mg bd or above were clinically efficacious and inhibited PARP-1 (Fong et al, 2009), so this combination was preferred over one with a higher dacarbazine dose and 20 mg bd olaparib. Although dose modifications were required for some patients, this dose was generally well tolerated in chemonaive melanoma patients, confirming its suitability for use outside of a clinical trial setting. Experience of myelosupression with PARP inhibitors and methylating agents to date has been variable. Use of AG-14699 required a reduction in the dose of temozolomide given concurrently, as was the case in the current study. However, full-dose temozolomide can be administered with a PARP-inhibitory dose of veliparib (ABT-888). The basis for this difference in the enhancement of myelotoxicity is not clear, as all the drugs are potent PARP-1 inhibitors.
The dose confirmation cohort was also designed to give a preliminary assessment of the efficacy of this dose combination, before potentially expanding recruitment to compare combination therapy with dacarbazine alone. The minimum efficacy requirement to move to the randomised phase of the trial was an objective response rate of 20% or greater after four cycles of treatment. As there were no further responses in the 10 melanoma patients treated first line with the combination, this requirement was not met and the expansion phase of the study was not opened. Progression-free survival was similar to that obtained with dacarbazine and temozolomide in previous clinical trials (Eigentler et al, 2003; Quirt et al, 2007).Given the limited numbers of chemotherapy-naive melanoma patients in this study, the possibility of a small benefit for the addition of the PARP inhibitor in this group of patients cannot be excluded.
The MPG activity was detectable in all samples analysed, showed considerable interpatient variation (Figure 2A) but was not significantly affected by administration of either olaparib or dacarbazine or sampling time (Figure 2B). It was anticipated that MPG activity might influence 7-meG levels, but this proved not to be the case (Figure 2C). As dacarbazine requires metabolic activation for conversion to a methylating species, interpatient differences in metabolism will influence initial methylation levels, potentially confounding any correlation between MPG activity and 7-meG. However, mean 7-meG levels measured 5 h after dacarbazine administration were remarkably consistent, so this is unlikely to explain the lack of correlation. Thus, in peripheral blood mononuclear cells, the availability of functional MPG is not the principal determining factor in the loss of 7-meG. Although there are no reports of other repair processes acting on this lesion, it is possible that factors such as MPG recruitment to damage site, cofactors and coupling to transcription-coupled or global genome repair processes, may modulate 7-meG removal.
A phase I study of AG014699 with temozolomide in patients with metastatic melanoma was the first clinical trial of a PARP inhibitor in combination with chemotherapy (Plummer et al, 2008). The combination was well tolerated, with some evidence of activity. However, in a phase II study of the combination, the haematological toxicity of temozolomide was exacerbated with one toxic death, three neutropaenic hospitalisations and dose reductions of temozolomide in a significant proportion of patients, highlighting the differences that can occur, as regimens identified in select populations in phase I trials are applied more widely. The level of PARP inhibitor activity has not been reported (Plummer et al, 2006). Other PARP inhibitors have been tested in melanoma in combination with a variety of cytotoxic or targeted agents. By far the largest study of this approach is the recently completed randomised phase 2 trial of ABT-888 and temozolomide. This trial of over 300 patients will provide the clearest insight into the potential for PARP inhibition in melanoma when its results become known late in 2010.
In conclusion, this phase I study identified clinically tolerable doses of olaparib and dacarbazine for use in combination therapy in patients with metastatic melanoma. However, no responses were seen in 10 chemotherapy-naive melanoma patients treated at the optimal combination doses identified. Despite the small size of this group, it is unlikely that this combination, at these doses, will provide a clinically important advantage over single-agent dacarbazine as first-line treatment in patients with advanced melanoma.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Bajetta E, Di Leo A, Zampino MG, Sertoli MR, Comella G, Barduagni M, Giannotti B, Queirolo P, Tribbia G, Bernengo MG (1994) Multicenter randomized trial of dacarbazine alone or in combination with two different doses and schedules of interferon alfa-2a in the treatment of advanced melanoma. J Clin Oncol 12: 806–811
Chalmers AJ (2009) The potential role and application of PARP inhibitors in cancer treatment. Br Med Bull 89: 23–40
Dantzer F, Schreiber V, Niedergang C, Trucco C, Flatter E, De La Rubia G, Oliver J, Rolli V, Menissier-de Murcia J, de Murcia G (1999) Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie 81: 69–75
Eigentler TK, Caroli UM, Radny P, Garbe C (2003) Palliative therapy of disseminated malignant melanoma: a systematic review of 41 randomised clinical trials. Lancet Oncol 4: 748–759
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS (2009) Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361: 123–134
Fromme JC, Banerjee A, Verdine GL (2004) DNA glycosylase recognition and catalysis. Curr Opin Struct Biol 14: 43–49
Harrison KL, Wood M, Lees NP, Hall CN, Margison GP, Povey AC (2001) Development and application of a sensitive and rapid immunoassay for the quantitation of N7-methyldeoxyguanosine in DNA samples. Chem Res Toxicol 14: 295–301
Menear KA, Adcock C, Boulter R, Cockcroft XL, Copsey L, Cranston A, Dillon KJ, Drzewiecki J, Garman S, Gomez S, Javaid H, Kerrigan F, Knights C, Lau A, Loh Jr VM, Matthews IT, Moore S, O’Connor MJ, Smith GC, Martin NM (2008) 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phth alazin-1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem 51: 6581–6591
Middleton MR, Grob JJ, Aaronson N, Fierlbeck G, Tilgen W, Seiter S, Gore M, Aamdal S, Cebon J, Coates A, Dreno B, Henz M, Schadendorf D, Kapp A, Weiss J, Fraass U, Statkevich P, Muller M, Thatcher N (2000) Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol 18: 158–166
Plummer R, Jones C, Middleton M, Wilson R, Evans J, Olsen A, Curtin N, Boddy A, McHugh P, Newell D, Harris A, Johnson P, Steinfeldt H, Dewji R, Wang D, Robson L, Calvert H (2008) Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res 14: 7917–7923
Plummer R, Lorigan P, Evans J, Steven N, Middleton M, Wilson R, Snow K, Dewji K, Calvert H (2006) First and final report of a phase II study of the poly(ADP-ribose) polymerase (PARP) inhibitor, AG014699, in combination with temozolomide (TMZ) in patients with metastatic malignant melanoma (MM). J Clin Oncol (Meeting Abstracts) 24: 8013
Quirt I, Verma S, Petrella T, Bak K, Charette M (2007) Temozolomide for the treatment of metastatic melanoma: a systematic review. Oncologist 12: 1114–1123
Tentori L, Portarena I, Vernole P, De Fabritiis P, Madaio R, Balduzzi A, Roy R, Bonmassar E, Graziani G (2001) Effects of single or split exposure of leukemic cells to temozolomide, combined with poly(ADP-ribose) polymerase inhibitors on cell growth, chromosomal aberrations and base excision repair components. Cancer Chemother Pharmacol 47: 361–369
Tentori L, Turriziani M, Franco D, Serafino A, Levati L, Roy R, Bonmassar E, Graziani G (1999) Treatment with temozolomide and poly(ADP-ribose) polymerase inhibitors induces early apoptosis and increases base excision repair gene transcripts in leukemic cells resistant to triazene compounds. Leukemia 13: 901–909
Tsuchida Y, Therasse P (2001) Response evaluation criteria in solid tumors (RECIST): new guidelines. Med Pediatr Oncol 37: 1–3
Virag L, Szabo C (2002) The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev 54: 375–429
Watson AJ, Middleton MR, McGown G, Thorncroft M, Ranson M, Hersey P, McArthur G, Davis ID, Thomson D, Beith J, Haydon A, Kefford R, Lorigan P, Mortimer P, Sabharwal A, Hayward O, Margison GP (2009) O(6)-methylguanine-DNA methyltransferase depletion and DNA damage in patients with melanoma treated with temozolomide alone or with lomeguatrib. Br J Cancer 100: 1250–1256
Acknowledgements
The trial was sponsored by Kudos Pharmaceuticals, a wholly owned subsidiary of AstraZeneca. MM and OK are supported by the NIHR Biomedical Research Centre, Oxford. MG acknowledges NHS funding to the NIHR Biomedical Research Centre at the Royal Marsden Hospital.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Khan, O., Gore, M., Lorigan, P. et al. A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. Br J Cancer 104, 750–755 (2011). https://doi.org/10.1038/bjc.2011.8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2011.8
Keywords
This article is cited by
-
Role of PARP1-mediated autophagy in EGFR-TKI resistance in non-small cell lung cancer
Scientific Reports (2020)
-
Phase I study of continuous olaparib capsule dosing in combination with carboplatin and/or paclitaxel (Part 1)
Investigational New Drugs (2020)
-
PARP Inhibitors and the Evolving Landscape of Ovarian Cancer Management: A Review
BioDrugs (2019)
-
PARP Inhibition in Cancer: An Update on Clinical Development
Targeted Oncology (2019)
-
Melanoma cells replicate through chemotherapy by reducing levels of key homologous recombination protein RAD51 and increasing expression of translesion synthesis DNA polymerase ζ
BMC Cancer (2017)
