
Abstract
The DNA damage response (DDR) has emerged as a critical tumour suppressor pathway responding to cellular DNA replicative stress downstream of aberrant oncogene over-expression. Recent studies have now implicated the DDR as a sensor of oncogenic virus infection. In this review, we discuss the mechanisms by which tumour viruses activate and also suppress the host DDR. The mechanism of tumour virus induction of the DDR is intrinsically linked to the need for these viruses to promote an S-phase environment to replicate their nucleic acid during infection. However, inappropriate expression of viral oncoproteins can also activate the DDR through various mechanisms including replicative stress, direct interaction with DDR components and induction of reactive oxygen species. Given the growth-suppressive consequences of activating the DDR, tumour viruses have also evolved mechanisms to attenuate these pathways. Aberrant expression of viral oncoproteins may therefore promote tumourigenesis through increased somatic mutation and aneuploidy due to DDR inactivation. This review will focus on the interplay between oncogenic viruses and the DDR with respect to cellular checkpoint control and transformation.
Similar content being viewed by others


Main
Innate tumour suppression in response to oncogenic stress includes the well-characterised ARF-mediated activation and stabilisation of p53 (Zindy et al, 2003; Christophorou et al, 2006; Efeyan et al, 2006) and the cellular DNA damage response (DDR) that is activated following oncogene-induced replicative stress (Halazonetis et al, 2008). As first recognised by Halazonetis, Bartek and colleagues, tumour cells often display an activated DDR as evidenced by foci of DDR signaling proteins such as 53BP1 and activated ATM (DiTullio et al, 2002). This work led to the landmark papers by Bartkova et al (2005) and Gorgoulis et al (2005), which demonstrated that acute over-expression of oncogenes caused replicative stress that was sensed by the ATR signaling pathway as well as double-stranded breaks recognised by the ATM pathway. Not long after the initial characterisation of these pathways, the functional significance of the DDR activation was revealed by genetic studies indicating that ATM and Chk2 were critical tumour suppressors downstream of oncogenes including H-RasV12, Mos, Cdc6, and cyclin E (Bartkova et al, 2006; Di Micco et al, 2006). Mechanistically, these data linked the well-studied DDR signalling pathway and known tumour suppressor functions of its components, including activation of checkpoints and p53-mediated apoptosis and senescence, to an oncogene-induced replicative stress. Although much of the initial work on the DDR pathway in tumour suppression has focused on cellular oncogenes, a recent body of literature indicates that viral oncogenes also engage the DDR.
Approximately, 20% of all cases of human cancer have an infectious aetiology, with ∼80% of those being viral (Bouvard et al, 2009) (Table 1). For example, Epstein-Barr virus (EBV) is nearly uniformly present in African Burkitt's lymphoma and AIDS-associated non-Hodgkin's lymphomas (Rickinson and Kieff, 2006). Kaposi's sarcoma-associated herpesvirus (KSHV) is responsible for Kaposi's sarcoma (KS) and primary effusion lymphomas commonly diagnosed in AIDS patients (Mesri et al, 2010). Furthermore, human papillomavirus (HPV) infection is central to the development of cervical cancer and is a major contributor to the global cancer burden (Moody and Laimins, 2010). Recently, several studies have revealed the tumour-suppressive role of the DDR in response to viral oncoproteins. A unique aspect of these interactions is the interplay between the virus and the host with respect to virus replication vs aberrant induction of growth control genes and inhibition of apoptosis. This review will focus on complex interactions between tumour viruses and the host DDR and outcomes that promote or prevent virus-induced tumourigenesis.
Viral oncoproteins provoke a tumour-suppressive DDR
The replication of tumour viruses is intrinsically linked to their ability to drive cell proliferation. Most of these viruses infect quiescent cells driving re-entry into the cell cycle to promote an environment conducive for viral nucleic acid replication. The consequences of such aberrant induction of cell proliferation include increased replicative stress, similar to that of cellular oncogene activation, leading to induction of the DDR. However, direct viral oncoprotein activation of the DDR also occurs through multiple mechanisms discussed below.
Tumour viruses activate the DDR by inducing cellular hyper-proliferation
Small DNA tumour viruses antagonise the transcriptionally repressive Rb family of proteins to promote E2F-driven cellular proliferation. Uncontrolled E2F activity has been shown to activate an ATM-dependant growth-suppressive DDR (Powers et al, 2004; Rogoff et al, 2004). HPV E7 and SV40 large T antigen are classic examples of viral oncoproteins targeting Rb by direct disruption of the interaction with E2F thereby increasing S-phase promoting E2F family members to drive cellular DNA replication (DeCaprio et al, 1988; Dyson et al, 1989; Munger et al, 1989; Cheng et al, 1995; Zalvide and DeCaprio, 1995). In recent work, E6 and E7 over-expression has been shown to induce replicative stress in primary keratinocytes suggesting that potent loss of growth control through E7-mediated Rb antagonism drives uncontrolled origin firing leading to damaged DNA that may contribute to cervical cancer pathogenesis (Bester et al, 2011). Similarly, SV40 large T antigen is sufficient to activate an ATM-induced DDR (Boichuk et al, 2010). However, as described later, SV40 large T antigen activates the DDR through multiple mechanisms including those independent of Rb interaction (Boichuk et al, 2010). Although the polyomavirus SV40 does not cause human cancer, the recently described Merkel cell polyomavirus (MCV or MCPyV) has been found clonally integrated in Merkel cell carcinomas (MCC) (Feng et al, 2008) and expresses a truncated large T antigen in these tumours lacking the capacity to replicate viral DNA (Shuda et al, 2008). These mutant large T antigens still retain the ability to perturb cell growth through Rb antagonism and likely activate the DDR providing selective pressure for mutations in DDR genes and downstream signalling leading to MCC.
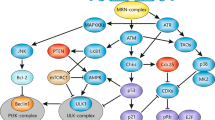
EBV infection of human B cells in vitro transiently activates an ATM-dependant DDR. EBV immortalises primary human B cells in culture mimicking physiological activation and survival signals, which when constitutively active is capable of driving B-cell lymphomas in vivo in the immune-suppressed. Recent work on EBV-infected primary human B cells indicates that early latent oncoprotein expression drives cellular hyperproliferation and activates ATM and downstream DDR checkpoints (Nikitin et al, 2010). Interestingly, inhibition of ATM and its downstream kinase Chk2 early in infection markedly increases the efficiency of transformation suggesting that the DDR blocks early events in EBV-mediated B-cell outgrowth. DDR activation by EBV correlates with heightened activity of the major viral trans-activator EBNA2 as measured by expression of EBNA2-dependant targets such as c-Myc and CD23. Both EBNA2 activity and DDR activation wane through infected cell divisions as EBNA2 transcriptional repressors, including EBNA3C, are activated. The genetic loss of EBNA3C, in fact, promotes an uncontrolled period of hyperproliferation that induces high-level ATM activation (Nikitin et al, 2010). Therefore, EBV has evolved to provoke a DDR owing to its need to drive B-cell proliferation, which is then limited by full expression of its latent viral oncoproteins in immortalised lymphoblastoid cell lines (Figure 1).

Interplay between viral oncoproteins and the host DDR. Viral oncoproteins activate cellular oncogenes (green arrows top level) in order to enter or re-enter the cell cycle, thereby inducing replicative stress and causing DNA single-stranded breaks (ssDNA). ssDNA and DNA double-stranded breaks (DSB) generated during repair of single-stranded DNA recognised by ATR and ATM kinases, respectively, which master regulate downstream signalling (all targets not shown), including activation of Chk2 and p53. Tumour virus oncoproteins modulate the function of DDR components by activating (green arrows) or suppressing (shown in red) their expression or activity.
Studies of the related γ-herpesvirus, KSHV, identified similar perturbations of the DDR signalling pathway. KSHV infects B cells and endothelial cells and can promote the development of primary effusion lymphomas and KS, two common cancers in AIDS patients (Mesri et al, 2010). Although the study of early events in KSHV de novo infection of primary cells has been limited, KSHV infection of immortalised endothelial cells in vitro induces the ATM signalling pathway (Koopal et al, 2007). Indeed, expression of the KSHV latent viral cyclin D homologue (v-cyclin) alone activates ATM. Moreover, investigation of KS tumours revealed activation of the DDR in early (patch), but not late (nodular), KS lesions (Koopal et al, 2007). Similar to EBV, elevated levels of DDR marks are likely induced by robust cellular proliferation. However, the downregulation of the DDR in advanced KS tumours is likely due to selection for mutations in the pathway allowing tumour cell survival.
Hepatitis B virus (HBV), which causes acute and chronic liver diseases, including cirrhosis and hepatocellular carcinoma, promotes cellular proliferation and the DDR through the pleiotropic oncoprotein HBx. Heterologous expression of HBx increases cytosolic Ca2+ levels leading to activation of Pyk2 and c-Src kinases (Klein and Schneider, 1997) and, ultimately, activation of Ras/Raf/MEK/ERK pathways. HBx expression can also promote p38MAPK pathway activation which upregulates E2F-dependant gene expression (Wang et al, 2008). Constitutive activation of these signalling pathways leads to activation of the ATR arm of the DDR pathway (Wang et al, 2008). The consequences of this activation, such as induction of S-phase arrest, are actually beneficial for virus replication despite being tumour suppressive (Zheng et al, 2011).
Direct viral protein activation of the ATM/Chk2 signaling pathway
Beyond the growth suppressive functions of the DDR, DNA repair and activation of checkpoints may be beneficial for the replication of tumour viral genomes. Oncogenic viruses have therefore developed mechanisms to activate specific components of the DDR pathway, while strictly preventing downstream induction of apoptosis. Recent work indicates that SV40 large T antigen can serve as both a substrate for the ATM kinase as well as its direct upstream activator through binding the Nbs1 component of the ATM-activating Mre11/Rad50/Nbs1 complex (Wu et al, 2004; Boichuk et al, 2010). ATM activation is actually necessary for viral DNA replication (Zhao et al, 2008b). However, as discussed below, the growth-suppressive consequences of ATM activation are attenuated downstream by large T antigen enabling SV40-infected cell survival.
HPV-infected cells display increased, but non-canonical ATM pathway activation. In particular, HPV oncoprotein-expressing undifferentiated keratinocytes display an activated DDR characterised by ATM, Chk1, Chk2, and H2AX phosphorylation (Moody and Laimins, 2009). However, upon differentiation of these cells, which increases virion genome replication, an additional set of ATM targets is phosphorylated including Nbs1 (Moody and Laimins, 2009). Interestingly, E7 was demonstrated to associate with the activated Ser1981-phoshorylated form of ATM independent of differentiation or other viral proteins (Moody and Laimins, 2009). Therefore, direct association between E7 and phospho-ATM, HPV episome amplification, and viral-induced replicative stress are all capable of activating the DDR and it remains unclear which of these activities is critical in regulating HPV pathogenesis (Moody and Laimins, 2009).
DDR activation through viral oncoprotein-mediated mitotic effects
Tumour viruses perturb normal cell cycle control in order to establish a constitutive S phase-like environment in which cellular factors are present required for viral replication. One consequence of this constitutive S-phase induction is inappropriate entry into mitosis, which activates DDR checkpoints including those triggered by Chk2 (Sato et al, 2010; Stolz et al, 2010). It was previously shown that KSHV v-cyclin expression promotes polyploidy and cytokinesis defects (Verschuren et al, 2002) and was subsequently confirmed by Ojala and colleagues that v-cyclin expression promotes amplification of centrosomes and intra-S-phase growth arrest (Koopal et al, 2007). Moreover, chemical inhibition of ATM/Chk2 led to aberrant mitoses and mitotic catastrophe in v-cyclin-expressing cells (Koopal et al, 2007).
In order to successfully transform cells, SV40 large T antigen targets the spindle assembly checkpoint component Bub1 leading to ATM/ATR activation (Cotsiki et al, 2004; Hein et al, 2009). Similarly, the high-risk HPV16 E6 and E7 proteins have been well documented to increase genomic instability by deregulating mitosis through the induction of multipolar spindles and centrosome duplication (Duensing et al, 2000). Specifically, E7 binding to nuclear mitotic apparatus protein 1 appears to deregulate normal chromosome alignment during prometaphase (Nguyen and Munger, 2009). More recently, E7 was observed to upregulate Polo-like kinase 4 (PLK4) expression leading to centriole multiplication (Korzeniewski et al, 2011). Therefore, multiple viral oncoproteins perturb mitosis through diverse mechanisms leading to an activated DDR.
Tumour viruses activate the DDR through induction of reactive oxygen species (ROS)
Elevated levels of reactive oxygen species can activate DDR pathways and may result in mutagenesis during oncogenic virus infection promoting tumourigenesis. Several tumour virus oncoproteins have been shown to increase ROS levels. For example, HTLV-1 Tax expression in fibroblasts or T cells induced a ROS-dependant DDR, although the mechanism by which ROS was induced remains unknown (Kinjo et al, 2010). Recently, Masucci and colleagues found that the EBV protein EBNA1 induced ROS levels and consequently ATM-dependant DDR activation and ultimately chromosomal aberrations (Gruhne et al, 2009a). Interestingly, EBNA1 induced ROS through upregulation of the mRNA encoding the catalytic subunit of the leukocyte NADPH oxidase NOX2, which directly promotes ROS accumulation (Gruhne et al, 2009a). A more recent study suggests that this EBNA1-driven ROS accumulation may promote telomere dysfunction, another known molecular signal for DDR activation (Kamranvar and Masucci, 2011). Given that EBNA1 is expressed in all EBV-positive tumour cells, its ability to induce ROS may promote tumourigenesis.
Viral proteins suppress the DDR to promote tumourigenesis
With the explicit purpose of providing an environment for virus replication, several tumour virus oncoproteins mitigate the growth-suppressive function of the DDR through altering downstream signalling events. However, the consequences of suppressing the DDR include aneuploidy and increased mutagenesis, which are major drivers of tumourigenesis. Tumour viruses have been well characterised to antagonise the function of the p53 tumour suppressor and more recently several viruses have been shown to target upstream checkpoint kinases as well.
Tumour virus suppression of downstream DDR signalling components
The small DNA tumour viruses SV40 and HPV have been well characterised for their ability to transform cells through perturbing activation of the DDR downstream target p53 (Kress et al, 1979; Lane and Crawford, 1979; Scheffner et al, 1990, 1993). This activity is thought to be a requirement for cell survival following aberrant S phase induction due to Rb antagonism by T Ag and E7 as described above. Although large DNA tumour viruses generally do not directly promote p53 degradation or abolish its function, the KSHV latent protein LANA and EBV latent protein EBNA3C have been shown to modulate p53 activity through direct association (Friborg et al, 1999; Yi et al, 2009; Chen et al, 2010). Other tumour viruses also directly antagonise p53 function including the HBV oncoprotein HBx, which both inhibits p53 DNA-binding activity and sequesters p53 in the cytoplasm thereby suppressing apoptosis (Wang et al, 1994, 1995; Elmore et al, 1997; Takada et al, 1997). HTLV-1 Tax suppresses p53 by directly antagonising its trans-activating function through both NFκB-dependant and NFκB-independent pathways (Ariumi et al, 2000; Pise-Masison et al, 2000; Miyazato et al, 2005). Although many tumour viral oncoproteins have been shown to associate with p53, the extents to which these activities contribute to pathogenesis remain unclear.
Viral oncoproteins directly target DDR checkpoint kinases
Upstream of p53 and cell cycle checkpoints are a series of DNA damage-sensing and signal-relaying kinases (Figure 1). Several viral oncoproteins directly target these upstream kinases through a number of mechanisms ultimately attenuating their function. For example, the HTLV-1 Tax oncoprotein directly binds to and inhibits signalling downstream of both Chk1 and Chk2 checkpoint kinases (Park et al, 2004, 2006; Gupta et al, 2007) as well as the upstream DNA damage-sensing DNA-PK (Durkin et al, 2008). Interestingly, Tax was also demonstrated to sequester the DDR components MDC1, DNA-PK and BRCA1 at artificial Tax-induced foci of pseudo-DNA damage as a unique mechanism to perturb endogenous DDR signalling pathways (Belgnaoui et al, 2010). Not unexpectedly, Tax expression attenuated ATM-downstream signalling leading to faster release of the G1/S checkpoint in response to ionising radiation (Chandhasin et al, 2008).
EBV attenuates DDR activity through indirect and also possibly direct mechanisms. As described above, at an early stage during EBV infection of primary B cells, an EBNA2-promoted, c-Myc-driven period of cellular hyperproliferation activates the DDR. Through subsequent cell divisions, the viral EBNA3C protein is expressed, which attenuates EBNA2 activity, hyperproliferation, and ultimately DDR activation (Nikitin et al, 2010). Therefore, as EBV-immortalised cells grow out in vitro, the DDR is no longer activated.
Under circumstances where EBV oncoproteins are aberrantly expressed, as evidenced in heterologous expression studies in EBV-negative B cells, DDR pathways can be directly attenuated. Specifically, Robertson and colleagues have observed a direct interaction between EBNA3C and Chk2 leading to decreased Chk2 activity, which may also contribute to DDR attenuation during primary B cell outgrowth (Choudhuri et al, 2007). Another study identified the latent membrane protein LMP1 as an inhibitor of ATM signalling due to transcriptional down-regulation of ATM upon LMP1 over-expression (Gruhne et al, 2009b). Under certain circumstances, such as in Hodgkin's lymphoma or nasopharyngeal carcinoma where LMP1 is expressed at high levels and may be important for cell survival, this activity may contribute to tumourigenesis due to the inability of ATM to trigger checkpoints and mediate efficient DNA repair.
Viral oncoproteins perturb mitotic checkpoint signalling
Mitotic checkpoints are often provoked by viral oncoprotein promotion of cell cycle progression. Therefore, in order for these viruses to replicate in the infected cell, signalling downstream of the G2/M checkpoint must be attenuated. Several oncogenic viruses encode proteins that precisely target this checkpoint with potentially catastrophic consequences on the karyotype of surviving cells. HTLV-1 Tax expression abolishes cellular mitotic checkpoints by directly targeting and prematurely activating the anaphase-promoting complex (Liu et al, 2005), as well as suppressing the spindle assembly checkpoint protein Mad 1 (Jin et al, 1998) resulting in highly aneuploid ATL cells. Similarly, the EBV EBNA3 proteins are capable of inhibiting the canonical G2/M checkpoint through suppression of p27 levels or activity depending on the cell type (Parker et al, 2000; Wade and Allday, 2000; Knight and Robertson, 2004). In addition, EBNA3C is capable of suppressing the effects of mitotic poisons in part through decreasing the levels of the spindle assembly checkpoint protein BubR1 (Leao et al, 2007; Gruhne et al, 2009b). The consequence of bypassing the mitotic checkpoint and DDR signalling downstream is the accumulation of aneuploid cells that can promote tumourigenesis through copy number amplification of oncogenes or loss of tumour suppressors.
Conclusions
In order to propagate their genomes, human tumour viruses induce robust cellular DNA replication that can lead to a replicative stress-activated host tumour-suppressive DDR. Although replicative stress is a common consequence of tumour virus infection, different strategies are used by these viruses to overcome the host DDR. Small DNA tumour viruses drive cellular DNA replication activating the host DDR, but also encode inhibitors of DDR downstream effectors such as HPV E6 or polyomavirus large T antigens that suppress p53. Activation of the host DDR induces S-phase arrest and allows such viruses to replicate in an S-phase like environment with the proper milieu of cellular replication factors. However, as p53 is a merging point of several tumour-suppressive pathways, its inhibition by viral proteins enables these viruses to avoid induction of p53-induced apoptosis or senescence. Inadvertent integration of viral genomes generates cells that produce DDR-provoking oncoproteins in the absence of viral replication. For example, MCV integrants express a truncated T antigen capable of Rb association, but not viral DNA replication (Feng et al, 2008). The consequences of such a scenario is the rapid proliferation of pre-malignant cells within which mutations in DDR components can be selected or, if still expressed, viral proteins can promote genomic instability by continuously inhibiting checkpoint function. It is these cells, which are far from the intentional output of the initial virus infection, that drive tumourigenesis.
The interplay between large DNA tumour viruses, such as EBV and KSHV, with the DDR is somewhat different. These viruses can replicate their episomes during the proliferation of latently infected host cells and only under specific circumstances (i.e. host cell differentiation) do they replicate genomes lytically to produce progeny virions. In order to ensure an environment that allows latent viral episome replication, viral oncoproteins promote S phase and cell proliferation. Similar to small DNA tumour viruses, these activities can lead to replicative stress and DDR activation. However, it is likely that with more complex genomes, the large DNA tumour viruses are able to attenuate the amount of replicative stress generated to maintain a successful latent infection. Such is the case for EBV, where an initial DDR-activating burst of proliferation is driven by the viral proteins EBNA2/EBNA-LP. Subsequently, expression of the EBNA3C protein attenuates EBNA2 transcriptional activity and DDR activation enabling long-term latency establishment (Nikitin et al, 2010). It is only in the inadvertent case when mutations in DDR components are generated by replicative stress and coupled with aberrantly over-expressed viral proteins such as EBV EBNA1 or EBNA3C or in the case of KSHV LANA or v-cyclin that these viruses might drive genomic instability and tumourigenesis. Importantly, this is not productive for the virus, but is rather a by-product of a highly evolved set of mechanisms aimed to drive cell proliferation enabling virus replication.
It is also worth noting that during lytic viral DNA replication, the nuclear sites of viral DNA synthesis often recruit DDR factors and also activate the DDR. These factors can either be beneficial for replication as is the case for ATM signalling in γ-herpesviruses (Tarakanova et al, 2007; Bouvard et al, 2009; Li et al, 2011) and papillomaviruses (Moody and Laimins, 2009) or detrimental as in the case of the ATM-activating Mre11/Rad50/Nbs1 proteins precluding processing of adenovirus DNA ends before packaging (Stracker et al, 2002). Therefore, the DDR can serve quite distinct functions depending on the molecular nature of the DNA damage and need for recombination and repair during genome replication.
In summary, the DDR can be activated directly by aberrant expression of oncoproteins, cellular or viral, or as a consequence of cellular proliferation-induced replicative stress. DNA tumour virus-driven cellular transformation occurs as a by-product of the virus promoting the cell cycle in order to establish an appropriate environment with the requisite DNA replication machinery and repair factors necessary for viral DNA replication. Similarly, viruses such as HTLV-1 must activate the infected T-cell in order to promote a favourable environment for proviral DNA integration. However, in the inadvertent setting such as following aberrant integration of viral genomes where loss of normal viral replication function occurs or other changes lead to increased viral oncoprotein expression, a constitutively activated DDR is triggered. DDR signalling typically limits viral oncogenesis, but also provides selective pressure for mutations in DDR signalling components that promote tumourigenesis. The delicate balance between virus replication, latency, and the extent of activation of the DDR ultimately dictates whether an infected cell will give rise to a productive cycle generating progeny virions or a tumour.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Anupam R, Datta A, Kesic M, Green-Church K, Shkriabai N, Kvaratskhelia M, Lairmore MD (2011) Human T-lymphotropic virus type 1 p30 interacts with REGgamma and modulates ATM (ataxia telangiectasia mutated) to promote cell survival. J Biol Chem 286 (9): 7661–7668
Ariumi Y, Kaida A, Lin JY, Hirota M, Masui O, Yamaoka S, Taya Y, Shimotohno K (2000) HTLV-1 tax oncoprotein represses the p53-mediated trans-activation function through coactivator CBP sequestration. Oncogene 19 (12): 1491–1499
Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J (2005) DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434 (7035): 864–870
Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, Takaoka M, Nakagawa H, Tort F, Fugger K, Johansson F, Sehested M, Andersen CL, Dyrskjot L, Orntoft T, Lukas J, Kittas C, Helleday T, Halazonetis TD, Bartek J, Gorgoulis VG (2006) Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444 (7119): 633–637
Belgnaoui SM, Fryrear KA, Nyalwidhe JO, Guo X, Semmes OJ (2010) The viral oncoprotein tax sequesters DNA damage response factors by tethering MDC1 to chromatin. J Biol Chem 285 (43): 32897–32905
Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B (2011) Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145 (3): 435–446
Boichuk S, Hu L, Hein J, Gjoerup OV (2010) Multiple DNA damage signaling and repair pathways deregulated by simian virus 40 large T antigen. J Virol 84 (16): 8007–8020
Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, Cogliano V (2009) A review of human carcinogens – part B: biological agents. Lancet Oncol 10 (4): 321–322
Chandhasin C, Ducu RI, Berkovich E, Kastan MB, Marriott SJ (2008) Human T-cell leukemia virus type 1 tax attenuates the ATM-mediated cellular DNA damage response. J Virol 82 (14): 6952–6961
Chen W, Hilton IB, Staudt MR, Burd CE, Dittmer DP (2010) Distinct p53, p53:LANA, and LANA complexes in Kaposi's Sarcoma--associated Herpesvirus Lymphomas. J Virol 84 (8): 3898–3908
Cheng S, Schmidt-Grimminger DC, Murant T, Broker TR, Chow LT (1995) Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes Dev 9 (19): 2335–2349
Choudhuri T, Verma SC, Lan K, Murakami M, Robertson ES (2007) The ATM/ATR signaling effector Chk2 is targeted by Epstein-Barr virus nuclear antigen 3C to release the G2/M cell cycle block. J Virol 81 (12): 6718–6730
Christophorou MA, Ringshausen I, Finch AJ, Swigart LB, Evan GI (2006) The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature 443 (7108): 214–217
Cotsiki M, Lock RL, Cheng Y, Williams GL, Zhao J, Perera D, Freire R, Entwistle A, Golemis EA, Roberts TM, Jat PS, Gjoerup OV (2004) Simian virus 40 large T antigen targets the spindle assembly checkpoint protein Bub1. Proc Natl Acad Sci USA 101 (4): 947–952
DeCaprio JA, Ludlow JW, Figge J, Shew JY, Huang CM, Lee WH, Marsilio E, Paucha E, Livingston DM (1988) SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell 54 (2): 275–283
Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, Maestro R, Pelicci PG, d’Adda di Fagagna F (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444 (7119): 638–642
DiTullio Jr RA, Mochan TA, Venere M, Bartkova J, Sehested M, Bartek J, Halazonetis TD (2002) 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat Cell Biol 4 (12): 998–1002
Duensing S, Lee LY, Duensing A, Basile J, Piboonniyom S, Gonzalez S, Crum CP, Munger K (2000) The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc Natl Acad Sci USA 97 (18): 10002–10007
Durkin SS, Guo X, Fryrear KA, Mihaylova VT, Gupta SK, Belgnaoui SM, Haoudi A, Kupfer GM, Semmes OJ (2008) HTLV-1 Tax oncoprotein subverts the cellular DNA damage response via binding to DNA-dependent protein kinase. J Biol Chem 283 (52): 36311–36320
Dyson N, Howley PM, Munger K, Harlow E (1989) The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243 (4893): 934–937
Efeyan A, Garcia-Cao I, Herranz D, Velasco-Miguel S, Serrano M (2006) Tumour biology: policing of oncogene activity by p53. Nature 443 (7108): 159
Elmore LW, Hancock AR, Chang SF, Wang XW, Chang S, Callahan CP, Geller DA, Will H, Harris CC (1997) Hepatitis B virus X protein and p53 tumor suppressor interactions in the modulation of apoptosis. Proc Natl Acad Sci USA 94 (26): 14707–14712
Feng H, Shuda M, Chang Y, Moore PS (2008) Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319 (5866): 1096–1100
Friborg Jr J, Kong W, Hottiger MO, Nabel GJ (1999) p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402 (6764): 889–894
Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio Jr RA, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD (2005) Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434 (7035): 907–913
Gruhne B, Sompallae R, Marescotti D, Kamranvar SA, Gastaldello S, Masucci MG (2009a) The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc Natl Acad Sci USA 106 (7): 2313–2318
Gruhne B, Sompallae R, Masucci MG (2009b) Three Epstein-Barr virus latency proteins independently promote genomic instability by inducing DNA damage, inhibiting DNA repair and inactivating cell cycle checkpoints. Oncogene 28 (45): 3997–4008
Gupta SK, Guo X, Durkin SS, Fryrear KF, Ward MD, Semmes OJ (2007) Human T-cell leukemia virus type 1 Tax oncoprotein prevents DNA damage-induced chromatin egress of hyperphosphorylated Chk2. J Biol Chem 282 (40): 29431–29440
Halazonetis TD, Gorgoulis VG, Bartek J (2008) An oncogene-induced DNA damage model for cancer development. Science 319 (5868): 1352–1355
Hein J, Boichuk S, Wu J, Cheng Y, Freire R, Jat PS, Roberts TM, Gjoerup OV (2009) Simian virus 40 large T antigen disrupts genome integrity and activates a DNA damage response via Bub1 binding. J Virol 83 (1): 117–127
Jin DY, Spencer F, Jeang KT (1998) Human T cell leukemia virus type 1 oncoprotein Tax targets the human mitotic checkpoint protein MAD1. Cell 93 (1): 81–91
Kamranvar SA, Masucci MG (2011) The Epstein-Barr virus nuclear antigen-1 promotes telomere dysfunction via induction of oxidative stress. Leukemia 25 (6): 1017–1025
Kinjo T, Ham-Terhune J, Peloponese Jr JM, Jeang KT (2010) Induction of reactive oxygen species by human T-cell leukemia virus type 1 tax correlates with DNA damage and expression of cellular senescence marker. J Virol 84 (10): 5431–5437
Klein NP, Schneider RJ (1997) Activation of Src family kinases by hepatitis B virus HBx protein and coupled signaling to Ras. Mol Cell Biol 17 (11): 6427–6436
Knight JS, Robertson ES (2004) Epstein-Barr virus nuclear antigen 3C regulates cyclin A/p27 complexes and enhances cyclin A-dependent kinase activity. J Virol 78 (4): 1981–1991
Koopal S, Furuhjelm JH, Jarviluoma A, Jaamaa S, Pyakurel P, Pussinen C, Wirzenius M, Biberfeld P, Alitalo K, Laiho M, Ojala PM (2007) Viral oncogene-induced DNA damage response is activated in Kaposi sarcoma tumorigenesis. PLoS Pathog 3 (9): 1348–1360
Korzeniewski N, Treat B, Duensing S (2011) The HPV-16 E7 oncoprotein induces centriole multiplication through deregulation of Polo-like kinase 4 expression. Mol Cancer 10: 61
Kress M, May E, Cassingena R, May P (1979) Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol 31 (2): 472–483
Lane DP, Crawford LV (1979) T antigen is bound to a host protein in SV40-transformed cells. Nature 278 (5701): 261–263
Leao M, Anderton E, Wade M, Meekings K, Allday MJ (2007) Epstein-barr virus-induced resistance to drugs that activate the mitotic spindle assembly checkpoint in Burkitt's lymphoma cells. J Virol 81 (1): 248–260
Li R, Zhu J, Xie Z, Liao G, Liu J, Chen MR, Hu S, Woodard C, Lin J, Taverna SD, Desai P, Ambinder RF, Hayward GS, Qian J, Zhu H, Hayward SD (2011) Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe 10 (4): 390–400
Liu B, Hong S, Tang Z, Yu H, Giam CZ (2005) HTLV-I Tax directly binds the Cdc20-associated anaphase-promoting complex and activates it ahead of schedule. Proc Natl Acad Sci USA 102 (1): 63–68
Liu J, Martin HJ, Liao G, Hayward SD (2007) The Kaposi's sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J Virol 81 (19): 10451–10459
Mesri EA, Cesarman E, Boshoff C (2010) Kaposi's sarcoma and its associated herpesvirus. Nat Rev Cancer 10 (10): 707–719
Miyazato A, Sheleg S, Iha H, Li Y, Jeang KT (2005) Evidence for NF-kappaB- and CBP-independent repression of p53's transcriptional activity by human T-cell leukemia virus type 1 Tax in mouse embryo and primary human fibroblasts. J Virol 79 (14): 9346–9350
Moody CA, Laimins LA (2009) Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog 5 (10): e1000605
Moody CA, Laimins LA (2010) Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer 10 (8): 550–560
Munger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM (1989) Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J 8 (13): 4099–4105
Nguyen CL, Munger K (2009) Human papillomavirus E7 protein deregulates mitosis via an association with nuclear mitotic apparatus protein 1. J Virol 83 (4): 1700–1707
Nikitin PA, Yan CM, Forte E, Bocedi A, Tourigny JP, White RE, Allday MJ, Patel A, Dave SS, Kim W, Hu K, Guo J, Tainter D, Rusyn E, Luftig MA (2010) An ATM/Chk2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human B cells. Cell Host Microbe 8 (6): 510–522
Park HU, Jeong JH, Chung JH, Brady JN (2004) Human T-cell leukemia virus type 1 Tax interacts with Chk1 and attenuates DNA-damage induced G2 arrest mediated by Chk1. Oncogene 23 (29): 4966–4974
Park HU, Jeong SJ, Jeong JH, Chung JH, Brady JN (2006) Human T-cell leukemia virus type 1 Tax attenuates gamma-irradiation-induced apoptosis through physical interaction with Chk2. Oncogene 25 (3): 438–447
Parker GA, Touitou R, Allday MJ (2000) Epstein-Barr virus EBNA3C can disrupt multiple cell cycle checkpoints and induce nuclear division divorced from cytokinesis. Oncogene 19 (5): 700–709
Pise-Masison CA, Mahieux R, Radonovich M, Jiang H, Duvall J, Guillerm C, Brady JN (2000) Insights into the molecular mechanism of p53 inhibition by HTLV type 1 Tax. AIDS Res Hum Retroviruses 16 (16): 1669–1675
Powers JT, Hong S, Mayhew CN, Rogers PM, Knudsen ES, Johnson DG (2004) E2F1 uses the ATM signaling pathway to induce p53 and Chk2 phosphorylation and apoptosis. Mol Cancer Res 2 (4): 203–214
Rickinson A, Kieff E (2006) Epstein-Barr virus. In Fields Virology, Knipe DM, Howley PM (eds) 5th edn, pp. 2603–2654. Lippincott, Williams, and Wilkins: Philadelphia
Rogoff HA, Pickering MT, Frame FM, Debatis ME, Sanchez Y, Jones S, Kowalik TF (2004) Apoptosis associated with deregulated E2F activity is dependent on E2F1 and Atm/Nbs1/Chk2. Mol Cell Biol 24 (7): 2968–2977
Sato K, Ohta T, Venkitaraman AR (2010) A mitotic role for the DNA damage-responsive CHK2 kinase. Nat Cell Biol 12 (5): 424–425
Scheffner M, Huibregtse JM, Vierstra RD, Howley PM (1993) The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75 (3): 495–505
Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM (1990) The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63 (6): 1129–1136
Shuda M, Feng H, Kwun HJ, Rosen ST, Gjoerup O, Moore PS, Chang Y (2008) T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci USA 105 (42): 16272–16277
Stolz A, Ertych N, Kienitz A, Vogel C, Schneider V, Fritz B, Jacob R, Dittmar G, Weichert W, Petersen I, Bastians H (2010) The CHK2-BRCA1 tumour suppressor pathway ensures chromosomal stability in human somatic cells. Nat Cell Biol 12 (5): 492–499
Stracker TH, Carson CT, Weitzman MD (2002) Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418 (6895): 348–352
Takada S, Kaneniwa N, Tsuchida N, Koike K (1997) Cytoplasmic retention of the p53 tumor suppressor gene product is observed in the hepatitis B virus X gene-transfected cells. Oncogene 15 (16): 1895–1901
Tarakanova VL, Leung-Pineda V, Hwang S, Yang CW, Matatall K, Basson M, Sun R, Piwnica-Worms H, Sleckman BP, Virgin IV HW (2007) Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 1 (4): 275–286
Verschuren EW, Klefstrom J, Evan GI, Jones N (2002) The oncogenic potential of Kaposi's sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in vivo. Cancer Cell 2 (3): 229–241
Wade M, Allday MJ (2000) Epstein-Barr virus suppresses a G(2)/M checkpoint activated by genotoxins. Mol Cell Biol 20 (4): 1344–1360
Wang WH, Hullinger RL, Andrisani OM (2008) Hepatitis B virus X protein via the p38MAPK pathway induces E2F1 release and ATR kinase activation mediating p53 apoptosis. J Biol Chem 283 (37): 25455–25467
Wang XW, Forrester K, Yeh H, Feitelson MA, Gu JR, Harris CC (1994) Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc Natl Acad Sci USA 91 (6): 2230–2234
Wang XW, Gibson MK, Vermeulen W, Yeh H, Forrester K, Sturzbecher HW, Hoeijmakers JH, Harris CC (1995) Abrogation of p53-induced apoptosis by the hepatitis B virus X gene. Cancer Res 55 (24): 6012–6016
Wu X, Avni D, Chiba T, Yan F, Zhao Q, Lin Y, Heng H, Livingston D (2004) SV40 T antigen interacts with Nbs1 to disrupt DNA replication control. Genes Dev 18 (11): 1305–1316
Yi F, Saha A, Murakami M, Kumar P, Knight JS, Cai Q, Choudhuri T, Robertson ES (2009) Epstein-Barr virus nuclear antigen 3C targets p53 and modulates its transcriptional and apoptotic activities. Virology 388 (2): 236–247
Zalvide J, DeCaprio JA (1995) Role of pRb-related proteins in simian virus 40 large-T-antigen-mediated transformation. Mol Cell Biol 15 (10): 5800–5810
Zhao F, Hou NB, Yang XL, He X, Liu Y, Zhang YH, Wei CW, Song T, Li L, Ma QJ, Zhong H (2008a) Ataxia telangiectasia-mutated-Rad3-related DNA damage checkpoint signaling pathway triggered by hepatitis B virus infection. World J Gastroenterol 14 (40): 6163–6170
Zhao X, Madden-Fuentes RJ, Lou BX, Pipas JM, Gerhardt J, Rigell CJ, Fanning E (2008b) Ataxia telangiectasia-mutated damage-signaling kinase- and proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in Simian virus 40-infected primate cells. J Virol 82 (11): 5316–5328
Zheng Z, Li J, Sun J, Song T, Wei C, Zhang Y, Rao G, Chen G, Li D, Yang G, Han B, Wei S, Cao C, Zhong H (2011) Inhibition of HBV replication by theophylline. Antiviral Res 89 (2): 149–155
Zindy F, Williams RT, Baudino TA, Rehg JE, Skapek SX, Cleveland JL, Roussel MF, Sherr CJ (2003) Arf tumor suppressor promoter monitors latent oncogenic signals in vivo. Proc Natl Acad Sci USA 100 (26): 15930–15935
Acknowledgements
We acknowledge the members of the Luftig laboratory for helpful discussion. This work was supported by grants from the Duke Center for AIDS Research (P30-AI064518), the National Institutes of Health (R01CA140337), Golfers Against Cancer, and the American Cancer Society.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Nikitin, P., Luftig, M. The DNA damage response in viral-induced cellular transformation. Br J Cancer 106, 429–435 (2012). https://doi.org/10.1038/bjc.2011.612
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2011.612
Keywords
This article is cited by
-
Human papillomavirus type 16 E6 and E7 oncoproteins interact with the nuclear p53-binding protein 1 in an in vitro reconstructed 3D epithelium: new insights for the virus-induced DNA damage response
Virology Journal (2018)
-
Detection of Merkel cell polyomavirus with a tumour-specific signature in non-small cell lung cancer
British Journal of Cancer (2013)
