
Abstract
Although tyrosine kinase inhibitors (TKIs) have significantly improved the prognosis of chronic myeloid leukemia (CML), the ability of TKIs to eradicate CML remains uncertain and patients must continue TKI therapy for indefinite periods. In this study, we performed whole-exome sequencing to identify somatic mutations in 24 patients with newly diagnosed chronic phase CML who were registered in the JALSG CML212 study. We identified 191 somatic mutations other than the BCR-ABL1 fusion gene (median 8, range 1–17). Age, hemoglobin concentration and white blood cell counts were correlated with the number of mutations. Patients with mutations ⩾6 showed higher rate of achieving major molecular response than those<6 (P=0.0381). Mutations in epigenetic regulator, ASXL1, TET2, TET3, KDM1A and MSH6 were found in 25% of patients. TET2 or TET3, AKT1 and RUNX1 were mutated in one patient each. ASXL1 was mutated within exon 12 in three cases. Mutated genes were significantly enriched with cell signaling and cell division pathways. Furthermore, DNA copy number analysis showed that 2 of 24 patients had uniparental disomy of chromosome 1p or 3q, which disappeared major molecular response was achieved. These mutations may play significant roles in CML pathogenesis in addition to the strong driver mutation BCR-ABL1.
Similar content being viewed by others


Introduction
Chronic myeloid leukemia (CML) is a clonal hematopoietic stem cell disorder characterized by a reciprocal translocation between the long arms of chromosomes 9 and 22, resulting in the production of the BCR-ABL1 fusion gene.1 Imatinib, a first-generation tyrosine kinase inhibitor (TKI), has significantly improved the prognosis of CML.2 Two second-generation TKIs, nilotinib and dasatinib have been recently approved as frontline treatments for newly diagnosed CML.3, 4 These two drugs are more effective than imatinib, and most patients achieve a faster and deeper molecular response than with imatinib.5, 6 However, the ability of TKIs to eradicate the CML clone remains uncertain; thus, CML patients may have to continue TKI therapy for indefinite periods. Therefore, a therapeutic goal is the discontinuation of TKIs and development of a curative treatment for CML.
The pathological status of myeloproliferative neoplasms (MPNs) is similar to that of CML because MPNs are also characterized by a very strong driver mutation of JAK2 V617F. Klampfl et al. examined somatic mutations of MPNs, essential thrombocythemia (ET), polycythemia vera (PV) and primary myelofibrosis (PMF) by whole-exome sequencing (WES) and identified mutations to MPL and CALR in addition to JAK2 V617F in ET and PMF.7 Another group reported the presence of somatic mutations in MPNs, with the JAK2 mutation being the most frequent, followed by the CALR mutation.8 In addition to JAK2 and CALR, somatic mutations were also identified in TET2, DNMT3A, ASXL1, and EZH2 in MPN patients. These genes are reported to be frequently mutated in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). Therefore, these mutations may have significant influences on the pathogenesis of MPNs.
The BCR-ABL1 fusion gene is a strong driver mutation in CML pathogenesis. However, there exists relatively few reports of somatic mutational analysis in CML. Therefore, the objective of the present study was to identify somatic mutations in patients with newly diagnosed CML in the chronic phase (CML-CP) by WES.
Materials and methods
Patients
The Japan Adult Leukemia Study Group (JALSG) CML212 study is a multicenter prospective randomized study to compare the cumulative achievement of CMR for adult de novo CML-CP (UMIN Clinical Trials Registry UMIN000007909, http://www.umin.ac.jp/ctrj/). Patients are randomized to either dasatinib or nilotinib. The primary endpoint of the study is a cumulative achievement of CMR by 18 months. Samples from the initial 24 patients enrolled in the JALSG CML212 study between May 2013 and Jan 2014 were analyzed in the present study. We obtained informed consent from all patients to use their samples for banking and molecular analysis, and approval was obtained from the ethics committees of the participating institutes, including the ethical committee of the Graduate School of Medicine, Chiba University (Approval No. 942).
Wes, deep sequencing and Sanger sequencing
WES and deep sequencing were performed as previously reported.9, 10, 11 Briefly, genomic DNA was extracted from peripheral blood mononuclear cells (PBMCs) at the time of CML diagnosis. As a germline control, DNA was obtained from buccal mucosal cells. PBMC DNA was also extracted when a patient achieved a major molecular response (MMR). Whole-exome capture was accomplished by liquid phase hybridization of sonicated genomic DNA with a mean length of 150–200 bp for the bait cRNA library, which was synthesized on magnetic beads (SureSelect Human ALL Exon V5; Agilent Technology, Santa Clara, CA, USA), according to the manufacturer’s protocol. The captured targets were subjected to massive sequencing using HiSeq 2000 sequencing system (Illumina, Inc., San Diego, CA, USA) with the pair end 100 bp read option, according to the manufacturer’s instructions.
Copy number analysis was performed using in—house pipeline (Shiozawa et al. in preparation), in which total copy number of bait regions and common SNPs and allele frequency of heterozygous single-nucleotide polymorphiisms (SNPs) in tumor samples, were used as the input data. The mean coverage of >95% of the target sequences was analyzed at an average depth of more than × 20 (Supplementary Figure S1).
Sanger sequencing against selected variants was performed to validate the mutations identified by WES. We designed the PCR primers to produce PCR products of approximately 1000 bp in length that contained the mutated regions. PCR products were sequenced using the Big Dye Terminator v1.1 cycle sequencing kit (Applied Biosystems, Foster City, CA, USA) and an ABI 3100 Genetic analyzer (Applied Biosystems). The data were analyzed using FinchTV software (Geospiza, Inc., Seattle, WA, USA).
Deep sequencing was also performed for some mutations detected by WES to determine their accurate allele frequencies. Regions containing candidate mutations were amplified from 10–100 ng of DNA samples and prepared for the generation of sequencing libraries using the SureSelectXT2 Reagent kit (Agilent Technologies) according to the manufacturer's instructions. The prepared library was subjected to deep sequencing using a Miseq sequencer (Illumina, Inc.). Obtained Fastq files from the MiSeq were analyzed for image analysis, base calling, and mapping using Strand NGS software (Strand Genomics, Inc., San Francisco, CA, USA). With Strand NGS, the sequencing quality score (−10 × log10 (P-value)) calculated with the Bayesian SNP calling algorithm of each base read was used for statistical analyses. For example, a quality score of 20 means an error probability of 1 in 100, and a score of 30 means an error probability of 1 in 1000. We considered bases with quality scores higher than 20 as statistically significant.
Statistical analyses
Single-nucleotide variants (SNVs) were extracted from whole-exome sequences as somatic mutations. All mutations were compared with published SNP data (dbSNP131, 1000 genome project and an in-house database). Known synonymous SNPs, or SNVs with P-values ⩾0.001 compared with the valiant allele frequency (VAF) of peripheral blood leukocytes and oral mucosa by the Fisher’s exact test were excluded from further analysis. We also used Fisher’s exact test to confirm statistical significance against the results of deep sequencing. Correlations between the number of mutations and clinical factors were identified by the Pearson product—moment correlation coefficient. Receiver operating curve (ROC) analysis was used to establish the number mutations as predicting factor of achieving MMR. Statistical significance between the number of mutations and achieving MMR was identified by Fisher’s exact test. These analyses were performed using EZR software (Saitama Medical Center, Jichi Medical University, Saitama, Japan), which is a graphical user interface for R (The R Foundation for Statistical Computing, Vienna, Austria).12 Gene ontology (GO) analysis was used to evaluate functional enrichment in GO terms among mutated genes detected by WES. Sequencing reads were aligned to the human genome reference (hg19) using the Shirokane 2 supercomputer system, and Fisher’s exact test was used to calculate the P-values of GO analysis.
Results
Patient characteristics
The patient characteristics are shown in Table 1. The median age of the patients (18 males and 6 females) at diagnosis was 54.5 years (range, 23–77 years). The mean white blood cell count was 96.2±127.7 × 109 cells per liter, and the mean hemoglobin concentration was 12.7±2.6 g dl−1. Two patients (8.3%) had additional mutations besides t(9;22) that were detected by the G-band staining method. The median international scale (IS) for %BCR-ABL1/ABL1 was 56.9±28.4%, excluding two patients with a minor BCR-ABL1 mutation. Two patients (No. 4 and 7) dropped out this trial because of side effects and patient’s reasons. Of the remaining 22 patients, 18 patients (82%) achieved MMR at 2 years, while four patients did not (Figure 2).
Summary of mutated genes
The WES results identified 191 somatic gene mutations in 24 patients (Supplementary Table S1). The number of mutations for each patient ranged from 1 to 17, with a median of 8. Correlation coefficients were calculated between the number of mutations and clinical factors, which revealed mild positive correlations with age (r=0.50, P<0.05) and hemoglobin concentration (r=0.48, P<0.05), with a moderate negative correlation with white blood cell count (r=−0.53, P<0.067). There were no correlations between the level of major BCR-ABL/ABL% IS (r=0.12), EUTOS score (r=−0.33), or Sokal score (r=−0.01) with the number of mutations. ROC analysis established 6 mutations as a cutoff level for comparing the rate of achieving MMR with an area under the curve of 0.69. Patients with mutations ⩾6 showed higher rate of achieving MMR (n=15 put of 17, 88.2%) than those<6 (3 out of 7, 42.9%; P=0.0381).
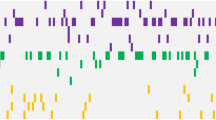
Among the 191 mutations, 166 were missense, which included four splice-site mutations. The remaining mutations were either frameshift (n=8) or non-frameshift (n=3) indels or nonsense (n=14). The types of somatic changes identified in each patient are shown in Figure 1.

Somatic mutations in CML-CP detected by WES. Number of validated somatic changes and the types of mutation in 24 patients are shown in different colors. Splice site mutations are shown in yellow, nonsense in red, indel in blue and missense in gray.
We found mutations in epigenetic regulator, ASXL1, TET2, TET3, KDM1A and MSH6 in 6 out of 24 patients (25%, Figure 2). ASXL1 was mutated in three cases, all of them were found within exon 12 (Figure 3). TET2, TET3, KDM1A and MSH6 were mutated in one case each. There were recurrent mutations expressed in CLSTN2, COL7A1, CSMD2 and DYSF. Recurrent mutations or mutations previously reported to be related with hematological malignancies such as RUNX1 or AKT1 were validated using Sanger sequencing or deep sequencing and summarized into 7 functional groups with DNA copy number alterations in Figure 2 and Table 2.

Summary of somatic mutations of interest and DNA copy number alterations detected by WES in 24 CML cases. Of all 191 somatic mutations identified by WES, recurrent mutations or mutations previously reported as being related to hematological malignancies were identified. Nonsense mutations are shown in red, missense in gray, frameshift insertions or deletions, and non-frameshift deletions in pink, blue, and purple, respectively. DNA copy number alterations were detected by WES, and UPDs are shown in green. The number on the cells describes the chromosome and its arm with UPD. Patients’ age at diagnosis and achievement of MMR are described in the last.

Mutations found in ASXL1 gene. ASXL1 exist on chromosome 20q11. Three mutations in ASXL1 were expressed in exon 12 showing with red arrows; all of them were loss-of-function mutations. The C-terminal of exon 12 contains a PHD finger that is a structural motif found in nuclear proteins and has reported to be involved in transcriptional regulation, chromatin modifications, and histone demethylation.
Copy number alterations by WES
DNA copy number alterations were analyzed by comparing the total copy number with the allele specific copy number detected by WES. Two patients (No. 7 and 17) were found to have uniparental disomy (UPD) of chromosomes 1p and 3q, respectively (Figure 4). SFPQ on 1p of patient #7 was mutated, while there was no mutation of 3q in patient No. 17. The VAF of mutated SFPQ was 0.47.

DNA copy number alteration in two cases with UPD, detected by WES. Patients No. 7 and 17 had UPD in chromosomes 1p and 3q, respectively. Copy number alterations were analyzed by comparing the total copy number with the allele—specific copy number. Blue dots on the upper line indicate the total copy number, while red and green dots on the lower line represent each the allele-specific copy number of hetero SNPs.
Heterozygous SNPs in ABCC5, C3orf37, IQCJ and PRR23A of chromosome 3q were discovered in buccal cells from patient No. 17; therefore, we performed Sanger sequencing with a DNA samples derived from PBMCs collected at diagnosis and after achieving MMR. The heterozygous SNPs of these four genes were all homozygously mutated at diagnosis and then returned to heterozygous mutations once MMR was achieved. These results suggest that UPD of chromosome 3q in patient No. 17 disappeared with TKI treatment.
Results of GO analysis
GO analysis, performed to evaluate functional enrichment in GO terms among mutated genes detected by WES, found that mutated genes were mostly enriched with cell signaling and cell division pathways. Among the results, GO terms with the P-value <0.01 and recurrently annotated were listed in Supplementary Table S2. Moreover, some GO terms thought to be related with tumorigenesis were selected on Figure 5.

Results of GO analysis. GO analysis was performed to evaluate functional enrichment in GO terms among mutated genes detected by WES. Sequencing reads were aligned to the human genome reference (hg19). GO terms with lower P values calculated using the Fisher’s exact test and higher frequency of annotation are shown.
Deep sequencing with MMR samples
Paired samples obtained from PBMCs both at diagnosis and after achieving MMR were available for three patients with mutated ASXL1 (patient No. 12), RUNX1 (patient No. 17), and KDM1A (patient No. 23) at diagnosis. We performed deep sequencing with these paired DNA samples to detect consecutive changes of mutations and found transition of VAF in these three mutations. These mutations of ASXL1, RUNX1 and KDM1A all disappeared once MMR was achieved (Supplementary Figure S2).
Discussion
We identified 191 somatic mutations, other than the BCR-ABL1 fusion gene, by WES in 24 newly diagnosed CML-CP patients. GO analysis revealed that the mutated genes were significantly enriched with cell signaling and cell division pathways. This result suggests that the cell signaling or cell division pathway was activated at CML onset. Some mutations in epigenetic regulator, ASXL1, TET2, TET3, KDM1A and MSH6 were found in 25% of patients. Moreover, AKT1, a kinase activator and RUNX1, a promotor of transcriptional regulation, have been reported as frequently mutated genes in AML/MDS and MPNs, and were mutated in one patient each.
DNA methylation has reported to be associated with pathogenesis of CML.13 Amabile et al. reported that aberrant DNA methylation of CML in murine models.14 DNA methylation changes were driven by BCR-ABL1 expression and contributed to the disease evolution. DNA methylation changes can act as a secondary event and contribute to leukemia formation, and using 5-Azacytidine, a DNA methyltransferase inhibitor, prolonged survival rate of murine model of CML. Moreover, methylation of additional genes other than BCR-ABL1 has reported in TKI-resistant CML patients, and is associated with their prognosis.15 Taken together, epigenetic regulation may play important roles for the pathology of CML.
Three loss-of-function mutations (frameshift insertion, deletion or nonsense mutation) found in ASXL1 all existed within exon 12. ASXL1 has well-known roles in histone modification and as a putative tumor suppressor gene that is often reported to be mutated in hematological malignancies. In MDS/MPN patients, ASXL1 mutations were concentrated within exon 12.16, 17 Moreover, Boultwood et al. performed sequencing analysis of ASXL1 within exon 12 of 41 pre-imatinib CML patients and identified ASXL1 mutations in six cases.18 Frameshift or nonsense mutations in exon 12 of ASXL1 should lead to the truncation of the protein and removal the C-terminal, which contains a PHD finger that is a structural motif found in nuclear proteins and has reported to be involved in transcriptional regulation, chromatin modifications and histone demethylation (Figure 5).17, 19, 20, 21, 22 A PHD finger recognizes the methylation status of histone lysine residues, such as histone H3 lysine 4, and its mutation has been reported in many diseases, including hematological malignancies.23 Taken together, the loss-of-function mutation of ASXL1 leads to PHD finger dysregulation, which may be related to tumorigenesis in CML. Moreover, deep sequencing revealed that the ASXL1 mutation of patient #12 at diagnosis had disappeared once MMR was achieved, while Boultwood et al. also reported ASXL1 mutations in four patients with CML-CP.18 These results suggest that an ASXL1 mutation indicate the disease state or prognosis of CML.
TET2 is one of the epigenetic regulator genes and frequently mutated in hematological malignancies, including CML.24 Wang et al. performed systematic mutation analysis by WES of PV and found recurrent somatic mutations in ASXL1, DNMT3A, TET2 and SF3B1.25 Ortmann et al.26 reported TET2 mutations in JAK2 mutation-positive MPN patients to clarify the effect of mutation order on disease phenotype and progression. They performed clonogenic analysis and detected, which patient is ‘TET2 first’ or ‘JAK2 first.’ The majority of bone marrow progenitor cells were common myeloid progenitors in ‘TET2 first’ patients, while megakaryocyte-erythroid progenitors were predominant in ‘JAK2 first’ patients. These reports suggest that the existence of another somatic mutation in addition to the strong driver mutation BCR-ABL1 may influence the disease phenotype. TET3 shares significant sequence homology with TET1 and TET2. Sequencing analysis of TET3 in myeloid malignancy, excluding CML, revealed no mutations among 96 myeloproliferative neoplasm patients.27 However, because TET2 and TET3 have overlapping requirements in hematopoietic stem cell emergence, the TET3 mutation may play a role in CML.28
KDM1A (also known as LSD1) is associated with the maintenance and differentiation of HSCs by demethylation of H3K4me2.29 KDM1A is upregulated in prostate cancer or neuroblastoma, and its expression has been reported to correlate with adverse outcome or inversely correlate with differentiation in tumors. KDM1A has been also reported as an essential regulator of leukemia stem cell potential in a murine model of human MLL-AF9 leukemia,30 with persistence of expression in associated oncogenic signaling, thereby preventing differentiation and apoptosis of leukemic cells. Moreover, treatment with the novel KDM1A antagonist significantly improved the survival of murine model of human AML, with inducing differentiation and apoptosis of leukemic cells.31 These results suggest that KDM1A may be related with maintenance of leukemic cell as an epigenetic regulator.
MSH6 is an essential component of the DNA mismatch repair mechanism and has been proposed to interact as an epigenetic regulator.32 Loss-of-function mutation was reported in relapsed ALL patients33 and the mutation leads to constitutional mismatch repair deficiency syndrome, which is characterized by the development of childhood cancers, mainly hematological malignancies.34 Taken together, epigenetic regulation may play important roles against the etiology of CML.
Recurrent somatic mutations in COL7A1, CSMD2, CLSTN2 and DYSF were also found in two patients each. It has been reported that COL7A1 expression was significantly upregulated in cancer stem cells in solid tumors by the positive stimulation of TGFB1 signaling.35 TGFB is a critical regulator of Akt activation in leukemia-initiating cells and controls FOXO3A localization in CML, which is responsible for maintaining leukemia-initiating cells.36 A CSMD2 mutation has not yet been reported in hematological malignancies, but it is a candidate tumor suppressor gene in pancreatic and colorectal cancers. Hypermethylation of CSMD2 in pancreatic cancer37 or its low expression in colorectal cancer was significantly associated with differentiation, lymphatic invasion, tumor size and overall survival.38 CLSTN2 encodes the synaptic protein calsyntenin 2 and is related to human memory and hippocampal function.39 DYSF is highly expressed in the skeletal muscle and has been suggested to be involved in membrane regeneration and repair. Recently, DYSF was also reported to be expressed in monocytes and its depletion impaired cell adhesion.40 However, mutations in these two genes DYSF are rarely reported in cancer.
Furthermore, there are several genes that have been previously reported in association with hematological malignancies. RUNX1 is a transcription factor that controls myeloid differentiation. Many reports have revealed that RUNX1 is mutated in the blastic crisis (BC) stage of CML (CML-BC), indicating that its mutation may affect CML progression.41, 42 RUNX1-deficient mice developed a mild myeloproliferative phenotype characterized by an increase in peripheral blood neutrophils, myeloid progenitor populations and extramedullary hematopoiesis.43 Furthermore, Zhao et al.44 reported transduction of both H78Q and V91fs—ter94 variants of RUNX1 into 32D cells or BCR-ABL-harboring murine cells, which resulted in disrupted myeloid differentiation and induction of a BC or accelerated phase-like phenotype in mice. These results suggest that RUNX1 alterations contribute to CML onset and progression.
In this study, copy number analysis by WES revealed UPD in chromosome 1p or 3q of two patients. Boultwood et al.18 also performed SNP array analysis of samples from 41 pre-imatinib CML-CP or -BC patients. A total of 65 regions of UPD were detected in 29 of 41 patients.
Eight recurrent regions of UPD were observed, and paired analysis of CP and BC samples identified two regions of UPD only in two patients in the BC phase. In our study, UPD on chromosome 3q in patient #17 disappeared with TKI treatment. Taken together, these data suggest that UPD is associated with disease evolution in CML. UPD could result from the mitotic recombination between chromatids of homologous chromosomes, which sometimes leads to transition from heterozygosity to homozygosity of each mutation. Mutations to JAK2 in PV,45 CBL in MPN,46 CEBPA in AML47 and RUNX1 in MDS/AML48 were found in association with UPD regions.
In this study, there were no significant mutations with UPD lesions on chromosome 1p and 3q, but there exist NRAS or JAK1 on chromosome 1p, while BCL6, GATA2 or TP63 on chromosome 3q. Some kinds of congenital disorders, such as Prader–Willi syndrome, are reported to have UPDs in certain imprinting regions.49 Dysfunction of imprinting genes caused by UPDs results in a disease onset. UPDs may affect the expression of genes.
In summary, we performed WES using samples collected from 24 newly diagnosed CML-CP patients. Although many recent studies have reported somatic mutations by next-generation sequencing in AML, MDS and MPN, this is the first report of somatic mutations in multiple cases of CML-CP. We found mutations of epigenetic regulator, ASXL1, TET2, TET3, KDM1A and MSH6 in 25% of patients, and also AKT1 and RUNX1 in each patient. Besides these mutations, multiple novel recurrent mutations previously reported in association with hematological malignancies were also found. Further analyses of long-term follow-up, functional analysis of these candidate genes or detecting transition of these mutations by deep sequencing may predict whether somatic mutations other than BCR-ABL1 can be related to their prognosis such as therapeutic resistance or relapse.
References
Apperley JF . Chronic myeloid leukaemia. Lancet 2015; 385: 1447–1459.
O'Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2003; 348: 994–1004.
Kantarjian H, Giles F, Wunderle L, Bhalla K, O'Brien S, Wassmann B et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med 2006; 354: 2542–2551.
Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med 2006; 354: 2531–2541.
Hughes TP, Saglio G, Kantarjian HM, Guilhot F, Niederwieser D, Rosti G et al. Early molecular response predicts outcomes in patients with chronic myeloid leukemia in chronic phase treated with frontline nilotinib or imatinib. Blood 2014; 123: 1353–1360.
Jabbour E, Kantarjian HM, Saglio G, Steegmann JL, Shah NP, Boque C et al. Early response with dasatinib or imatinib in chronic myeloid leukemia: 3-year follow-up from a randomized phase 3 trial (DASISION). Blood 2014; 123: 494–500.
Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013; 369: 2379–2390.
Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013; 369: 2391–2405.
Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011; 478: 64–69.
Yoshida K, Toki T, Okuno Y, Kanezaki R, Shiraishi Y, Sato-Otsubo A et al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat Genet 2013; 45: 1293–1299.
Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yoshida K, Townsley D et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med 2015; 373: 35–47.
Kanda Y . Investigation of the freely available easy-to-use software 'EZR' for medical statistics. Bone Marrow Transplant 2013; 48: 452–458.
Machova Polakova K, Koblihova J, Stopka T . Role of epigenetics in chronic myeloid leukemia. Curr Hematol Malig Rep 2013; 8: 28–36.
Amabile G, Di Ruscio A, Muller F, Welner RS, Yang H, Ebralidze AK et al. Dissecting the role of aberrant DNA methylation in human leukaemia. Nat Commun 2015; 6: 7091.
Dunwell T, Hesson L, Rauch TA, Wang L, Clark RE, Dallol A et al. A genome-wide screen identifies frequently methylated genes in haematological and epithelial cancers. Mol Cancer 2010; 9: 44.
Carbuccia N, Murati A, Trouplin V, Brecqueville M, Adelaide J, Rey J et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia 2009; 23: 2183–2186.
Gelsi-Boyer V, Trouplin V, Adelaide J, Bonansea J, Cervera N, Carbuccia N et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol 2009; 145: 788–800.
Boultwood J, Perry J, Zaman R, Fernandez-Santamaria C, Littlewood T, Kusec R et al. High-density single nucleotide polymorphism array analysis and ASXL1 gene mutation screening in chronic myeloid leukemia during disease progression. Leukemia 2010; 24: 1139–1145.
Bienz M . The PHD finger, a nuclear protein-interaction domain. Trends Biochem Sci 2006; 31: 35–40.
Cloos PA, Christensen J, Agger K, Helin K . Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev 2008; 22: 1115–1140.
Abdel-Wahab O, Pardanani A, Patel J, Wadleigh M, Lasho T, Heguy A et al. Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia 2011; 25: 1200–1202.
Katoh M . Functional and cancer genomics of ASXL family members. Br J Cancer 2013; 109: 299–306.
Baker LA, Allis CD, Wang GG . PHD fingers in human diseases: disorders arising from misinterpreting epigenetic marks. Mutat Res 2008; 647: 3–12.
Schmidt M, Rinke J, Schafer V, Schnittger S, Kohlmann A, Obstfelder E et al. Molecular-defined clonal evolution in patients with chronic myeloid leukemia independent of the BCR-ABL status. Leukemia 2014; 28: 2292–2299.
Wang L, Swierczek SI, Drummond J, Hickman K, Kim SJ, Walker K et al. Whole-exome sequencing of polycythemia vera revealed novel driver genes and somatic mutation shared by T cells and granulocytes. Leukemia 2014; 28: 935–938.
Ortmann CA, Kent DG, Nangalia J, Silber Y, Wedge DC, Grinfeld J et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med 2015; 372: 601–612.
Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009; 114: 144–147.
Li C, Lan Y, Schwartz-Orbach L, Korol E, Tahiliani M, Evans T et al. Overlapping requirements for Tet2 and Tet3 in normal development and hematopoietic stem cell emergence. Cell reports 2015; 12: 1133–1143.
Kerenyi MA, Shao Z, Hsu YJ, Guo G, Luc S, O'Brien K et al. Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. Elife 2013; 2: e00633.
Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 2012; 21: 473–487.
Fiskus W, Sharma S, Shah B, Portier BP, Devaraj SG, Liu K et al. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia 2014; 28: 2155–2164.
Li F, Mao G, Tong D, Huang J, Gu L, Yang W et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 2013; 153: 590–600.
Mar BG, Bullinger LB, McLean KM, Grauman PV, Harris MH, Stevenson K et al. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat Commun 2014; 5: 3469.
Wimmer K, Etzler J . Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg? Hum Genet 2008; 124: 105–122.
Oktem G, Sercan O, Guven U, Uslu R, Uysal A, Goksel G et al. Cancer stem cell differentiation: TGFbeta1 and versican may trigger molecules for the organization of tumor spheroids. Oncol Rep 2014; 32: 641–649.
Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y et al. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010; 463: 676–680.
Shimizu H, Horii A, Sunamura M, Motoi F, Egawa S, Unno M et al. Identification of epigenetically silenced genes in human pancreatic cancer by a novel method 'microarray coupled with methyl-CpG targeted transcriptional activation' (MeTA-array). Biochem Biophys Res Commun 2011; 411: 162–167.
Zhang R, Song C . Loss of CSMD1 or 2 may contribute to the poor prognosis of colorectal cancer patients. Tumour Biol 2014; 35: 4419–4423.
Jacobsen LK, Picciotto MR, Heath CJ, Mencl WE, Gelernter J . Allelic variation of calsyntenin 2 (CLSTN2) modulates the impact of developmental tobacco smoke exposure on mnemonic processing in adolescents. Biol Psychiatry 2009; 65: 671–679.
de Morree A, Flix B, Bagaric I, Wang J, van den Boogaard M, Grand Moursel L et al. Dysferlin regulates cell adhesion in human monocytes. J Biol Chem 2013; 288: 14147–14157.
Roche-Lestienne C, Deluche L, Corm S, Tigaud I, Joha S, Philippe N et al. RUNX1 DNA-binding mutations and RUNX1-PRDM16 cryptic fusions in BCR-ABL+ leukemias are frequently associated with secondary trisomy 21 and may contribute to clonal evolution and imatinib resistance. Blood 2008; 111: 3735–3741.
Soverini S, de Benedittis C, Mancini M, Martinelli G . Mutations in the BCR-ABL1 kinase domain and elsewhere in chronic myeloid leukemia. Clin Lymphoma Myeloma Leuk 2015; 15 (Suppl): S120–S128.
Growney JD, Shigematsu H, Li Z, Lee BH, Adelsperger J, Rowan R et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood 2005; 106: 494–504.
Zhao LJ, Wang YY, Li G, Ma LY, Xiong SM, Weng XQ et al. Functional features of RUNX1 mutants in acute transformation of chronic myeloid leukemia and their contribution to inducing murine full-blown leukemia. Blood 2012; 119: 2873–2882.
Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352: 1779–1790.
Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature 2009; 460: 904–908.
Snaddon J, Smith ML, Neat M, Cambal-Parrales M, Dixon-McIver A, Arch R et al. Mutations of CEBPA in acute myeloid leukemia FAB types M1 and M2. Genes Chromosomes Cancer 2003; 37: 72–78.
Flach J, Dicker F, Schnittger S, Schindela S, Kohlmann A, Haferlach T et al. An accumulation of cytogenetic and molecular genetic events characterizes the progression from MDS to secondary AML: an analysis of 38 paired samples analyzed by cytogenetics, molecular mutation analysis and SNP microarray profiling. Leukemia 2011; 25: 713–718.
Gunay-Aygun M, Schwartz S, Heeger S, O'Riordan MA, Cassidy SB . The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics 2001; 108: E92.
Acknowledgements
We thank Professor Osamu Ohara, Future Medicine Education and Research Organization at Chiba University for advice on experiments and technical assistances. This study was performed as a research program of the Project for Development of Innovative Research on Cancer Therapeutics (P-Direct), Ministry of Education, Culture, Sports, Science and Technology of Japan (No. 15ck0106189h0001), and also supported by the Practical Research for Innovative Cancer Control from Japan Agency for Medical Research and Development (AMED), and the National Cancer Center Research and Development Fund (26-A-24).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Blood Cancer Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Togasaki, E., Takeda, J., Yoshida, K. et al. Frequent somatic mutations in epigenetic regulators in newly diagnosed chronic myeloid leukemia. Blood Cancer Journal 7, e559 (2017). https://doi.org/10.1038/bcj.2017.36
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2017.36
This article is cited by
-
Whole-exome sequencing identified recurrent and novel variants in benzene-induced leukemia
BMC Medical Genomics (2023)
-
The importance of personalized medicine in chronic myeloid leukemia management: a narrative review
Egyptian Journal of Medical Human Genetics (2023)
-
Epigenetic regulation in hematopoiesis and its implications in the targeted therapy of hematologic malignancies
Signal Transduction and Targeted Therapy (2023)
-
Genetic landscape of chronic myeloid leukemia
International Journal of Hematology (2023)
-
Increased copy number of imprinted genes in the chromosomal region 20q11-q13.32 is associated with resistance to antitumor agents in cancer cell lines
Clinical Epigenetics (2022)
