
Abstract
Human autosomal recessive osteopetrosis (ARO), also known as infantile malignant osteopetrosis, is a rare genetic bone disorder that often causes death. Mutations in T-cell immune regulator 1 (TCIRG1) are a frequent cause of human ARO. Six additional genes (TNFSF11, TNFRSF11A, CLCN7, OSTM1, SNX10, PLEKHM1) were also found to be associated with human ARO. In order to expand the mutation spectrum and clinical diversity for a better understanding of the ARO phenotype and to further investigate the clinical characteristics of benign subjects with ARO, we here report five individuals with ARO from four unrelated Chinese families. X-ray examination was conducted and bone turnover markers were assayed. The gene of T-cell immune regulator 1 (TCIRG1) was screened and analyzed. Monocyte-induced osteoclasts were prepared and their resorption ability was studied in vitro. We identified five novel mutations (c.66delC, c.1020+1_1020+5dup, c.2181C>A, c.2236+6T>G, c.692delA) in these patients. Four patients displayed a malignant phenotype, three of them died, and one who received bone marrow transplantation survived. The remaining one, a 24-year-old male from a consanguineous family, was diagnosed based on radiological findings but presented no neurological or hematological defects. He was homozygous for c.2236+6T>G in intron 18; this mutation influenced the splicing process. An in vitro functional study of this novel splicing defect showed no resorption pits on dentine slices. TCIRG1-dependent osteopetrosis with a mild clinical course was observed for the first time in Chinese population. The present findings add to the wide range of phenotypes of Chinese patients with TCIRG1-dependent ARO and enrich the database of TCIRG1 mutations.
Similar content being viewed by others

Introduction
Human autosomal recessive osteopetrosis (ARO) represents a group of inherited bone disorders characterized by diffusely increased bone density due to the failure of bone resorption by osteoclasts1. ARO is a rare disease that has an incidence of 1 in 250 000 births2, but the incidence is particularly high in specific geographic regions (eg, Costa Rica, the Middle East, the Chuvash Republic of Russia and the Province of Västerbotten in northern Sweden). This distribution is attributed to the founder effect, geographic isolation or high parental consanguinity3. ARO renders bones more susceptible to hematological impairment and secondary neurological deficit (blindness or deafness), which are caused by a decreased bone marrow cavity and nerve compression. Li et al showed that the targeted disruption of Atp6i in mice resulted in severe osteopetrosis4. Then, in 2000, Frattini et al elucidated for the first time that mutations in T-cell immune regulator 1 (TCIRG1) are a frequent cause of human ARO5. Subsequently, molecular analysis has revealed that six additional genes (TNFSF11, TNFRSF11A, CLCN7, OSTM1, SNX10, PLEKHM1) are associated with human ARO. Mutations in TCIRG1 are responsible for more than 50% of ARO-affected individuals6, demonstrating the crucial role of the V-ATPase in osteoclast function. Despite rapid development in the understanding of the pathogenesis of osteopetrotic conditions, the genetic basis of approximately 30% of cases remains to be elucidated2.
Sobacchi et al and others have provided insight on a wide variety of TCIRG1 mutations, including missense, nonsense, small deletions/insertions, splice-site mutations, large genomic deletions and intronic mutations5,7,8,9,10. However, data concerning Chinese TCIRG1-deficient ARO patients are relatively limited, and include mainly reports of individual mutations11,12. Here, we described a relatively large sample of affected Chinese individuals with variable clinical severities, and we focused on the identification of the molecular defect and osteoclast functional characterization of a 24-year-old male who came from a consanguineous family. Moreover, the mild phenotype of osteopetrosis was observed for the first time in Chinese population. To date, the only cure is hematopoietic stem cell transplantation (HSCT), and we sincerely hope that more patients are reported and further studies are performed.
Materials and methods
Subjects and samples
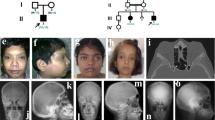
This study was approved by the Ethics Committee of the Shanghai Jiao Tong University Affiliated Sixth People's Hospital. The pedigrees of these families are shown in Figure 1, and the major clinical findings are summarized in Table 1.

(A) Pedigrees of the four Chinese families with TCIRG1-dependent ARO. Arrows indicate the probands. Filled black symbols refer to patients with ARO. Half-black symbols represent healthy carriers with a heterozygous mutation. (B) Location and sequencing of pathogenic TCIRG1 mutations. Family 1: c.1020+1_1020+5dup and c.66delc; Family 2: c.1213G>A and c.2181C>A; Family 3: c.292delA and c.909C>A; Family 4: c.2236+6T>G; (C). p.Tyr23ThrfsX4 (Y23), p.Lys231ArgfsX48 (K231), p.Gly405Arg (G405), p.Cys727X (C727) occur at a highly conserved position in TCIRG1, as shown by a comparison the corresponding sequence of 7 vertebrates.
Family 1
A 10-month-old proband (Pt 1, II1) (Figure 1A) was referred to us for genetic diagnosis with clinical manifestations of ARO. He was the only child of unrelated parents and was born at full-term via caesarean section. His tooth did not erupt when he was referred to us, and he was smaller than his contemporaries. His height was 62 cm (normal range: 71.0–76.3 cm) and weight was 7.0 kg (normal range: 8.6–10.6 kg). At approximately 9 months old, he suffered from acute infection with high fever, and routine blood tests showed clearly elevated leucocytes and decreased red cells, platelets and hemoglobin. Abdominal ultrasonography detected an enlarged spleen and liver. His liver margin was 4.0 cm below the right costae and the spleen margin was 6 cm below the left costae. Chest radiography confirmed generalized osteopetrosis, with a widened growth plate widened and nodular rib ends. Magnetic resonance imaging (MRI) showed hydrocephalus as well as a downward displacement of the cerebellum. After carefully evaluating the potential risks, surgeons successfully treated the patient with a ventriculo-peritoneal shunt for hydrocephalus. The patient received bone marrow transplantation (BMT) from two HLA-antigen mismatched related donors at approximately 13 months old. A combination of fludarabine, busulfan and cyclophosphamide was used as the conditioning regimen; he received approximately 29.9×108/kg mononuclear cells and 10.9×106/kg CD34+ cells. His illness was complicated by concomitant acute graft-versus-host-disease (GVHD), which was accompanied by diarrhea and skin rash. He is alive as of the time of this report.
Family 2
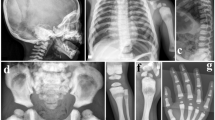
Patients 2A and 2B (Pt 2A, II1; Pt 2B, II2) (Figure 1A) were two deceased, affected siblings born from a non-consanguineous family. Pt 2A, a male, was born via natural childbirth and was suffering from fever, cough, nasal congestion and runny nose. He was admitted to a local hospital at approximately 20 d old; chest X-ray revealed typical rickets-like nodular costochondral junctions at the ends of the ribs and a widened growth plate. Further inspection confirmed osteopetrosis (Figure 2A). Blood tests indicated decreased serum calcium and phosphorus. Ultrasonography revealed that his liver was 24 cm below his right costae. Pathological features, such as overall growth retardation, delayed tooth eruption, tooth loss, poor vision and recurrent infections, became evident, and finally he died of multiple organ failure at 4 years old. His height was 92.0 cm (normal range: 98.7–107.2 cm) and weight was 12.2 kg (normal range: 14.8–18.7 kg). The patient had only four teeth at the time of death. Pt 2B, who was two years younger than Pt 2A, experienced a normal childbirth. At approximately one month old, his anxious parents took him to the hospital for physical examination. Radiographs revealed elevated bone mineral density. He suffered from growth retardation [height 75 cm (normal range: 84.3–91.0 cm) and decreased weight 10.5 kg (normal range: 11.2–14.0 kg) at two years old], anemia, and recurrent infections. His first tooth erupted at approximately one year old. He could not walk. He died of bone marrow failure at 2 years old.

Representative X-rays of Pt 2A, Pt 3, Pt 4, showing the diffusely increased bone density. (A) Pt 2A at 2 years, notice the increased vertebra and rib density as well as typical rickets-like nodular costochondral junctions at the end of ribs; (B) Pt 3 at 6 months, radiograph of the legs show bone sclerosis along with fraying and splaying of metaphyses, growth plate widening and diffuse periosteal reaction (arrows indicated); C, D, E, F, G are pictures of Pt 4. (C) The pelvic bones, as well as femoral head, proximal femora demonstrate high bone density; (D) Lateral cephalogram indicates sclerotic skull base and cervical vertebra; (E) Unlike the typical “rugger-jersey appearance” of CLCN7-dependent ADO type, this standard radiograph of the spine (anteroposterior) showing the homogeneous sclerosis of the vertebra; (F) Broken bone and external fixation when Pt 4 was 16 years old; (G) The obliteration of marrow cavity and bone deformity of right distal femur resulting from non-perfect healing and fracture remodeling.
Family 3
Patient 3 (Pt 3, II1, Figure 1A), a male, died of bone marrow failure at approximately 7 months old. His parents came to us for genetic counseling with a small piece of dried umbilical cord 15 months after his death. The proband was born to a non-consanguineous couple after an uneventful delivery, but he was admitted to a local hospital due to convulsion at 6 months. Blood tests revealed decreased serum calcium and hemoglobin. Total body radiographs (Figure 2B) confirmed bone sclerosis with a widened growth plate. During hospitalization, he developed bronchopneumonia, which worsened his outcome. No further inspections, such as abdominal ultrasonography or optic nerve computerized tomographic (CT) scanning, were performed before he died.
Family 4
A 24-year-old proband (Pt 4, IV2) (Figure 1A) was the second child of unaffected first-cousin parents. His older sister was healthy. His past medical history included tooth decay (eighteen years old, three molars of the right maxillary) and dental repair. He was initially evaluated at 16 years old because of a right distal femur fracture from jumping on the ground. The X-ray demonstrated a broken bone with an almost vanished medullary cavity because of sclerotic bone change (Figure 2C-2G). After the fracture healed, he did not receive any specific therapy, attended school as usual, avoided physical activity, and did not experience fracture, hematological or neurological defects. At the age of 24, he arrived at our clinic with back pain; his height was 159 cm and weight was 50.8 kg. Peripheral blood cells, calcium, phosphorus, alkaline phosphatase, parathyroid hormone, and 25-hydroxy vitamin D levels were normal. However, bone turnover markers, such as osteocalcin and beta C-terminal cross-linked telopeptides of type I collagen (β-CTX), were slightly elevated based on the reference ranges previously established13 (Table 1). The Z scores of lumbar 1-4, femoral neck and total hip were +16.6, +18.4, and +16.6, respectively. A thorough examination of the 3-generation pedigree excluded any disease symptoms in relatives. Currently, the patient is still in follow-up.
Molecular studies
DNA was extracted from peripheral white blood cells by proteinase K digestion followed by purification with phenol/chloroform and isopropyl alcohol precipitation. The DNA of Pt 3 was extracted from his dried umbilical cord using the QIAamp DNA Investigator kit (Qiagen, Hilden, Germany).
Molecular analysis was performed by amplification and direct sequencing of exons and intron-exon boundaries as previously described14. The sequences of the TCIRG1 primers used are available in Supplementary Table 1. After the peripheral blood mononuclear cells of Pt 4 were lysed in TRIzol, mRNA was isolated and reverse transcribed. The effect of intronic variants on TCIRG1 transcript processing was investigated at the cDNA level with the forward primer 5´-CCTACACGACGCTCTTCCGATCTAAAACAAGGCCGGGTTGCTG-3´ located in exon 17 and the reverse primer 5´-TCAGACGTGTGCTCTTCCGATCTAAGGCTGAGAGTCCCTCCATCAC-3´ in exon 19 (NM_006019.3) using the following thermocycling conditions: initial denaturation step at 95 °C for 2 min, followed by 40 cycles of denaturation at 95 °C for 15 s, annealing at 60 °C for 1 min and finally a dissociation stage. Paired-end sequencing was performed on the Illumina miseq2*250 platform with a sequencing depth of 5000×. The minimum data collected were 0.2 M.
Whole exome sequencing of Pt 4
We sequenced the exome of the single affected individual (Figure 1A, family 4, IV2) from a consanguineous family. Exon-enriched DNA from the proband was sequenced by the Illumina Genome Analyzer II platform according to the manufacturer's instructions. Raw image files were processed with Illumina Base Calling Software 1.7 with default parameters, and the sequences of each individual were reported as 90 bp paired-end reads. Sequence reads were mapped to a reference genome (UCSC Genome Brower hg18 assembly) with SOAP2 (BGI-Shenzhen)15,16,17. The SOAP SNP results were filtered according to a standard workflow for exome sequencing.
Analysis of in vitro osteoclast function of Pt 4
Peripheral blood mononuclear cells (PBMNCs) were isolated from 10 mL of blood from Pt 4 and an age-matched healthy male according to Lympholyte®-H (Cedarlane Laboratories Ltd) instructions. Then, PBMNCs were diluted with 1-2 mL α-Minimum Essential Medium (MEM; 10% fetal calf serum, 2 mmol/L L-glutamine, 100 U/mL penicillin, 0.1 mg/mL streptomycin) containing 20 ng/mL recombinant human macrophage colony-stimulating factor (rhM-CSF; R&D Systems) and seeded in 96-well plates and 6-well plates (2×105 and 2.5×106 cells/well, respectively) for 9 d, with a complete change of culture medium every 3 d. Then, M-CSF-dependent macrophages in 6-well plates were rinsed with cold PBS and scraped into fresh medium after incubation with 0.1% (1 mg/mL) trypsin for 15 min. After the cells were centrifuged (300×g, 5 min) and resuspended in fresh medium, they were seeded onto 96-well plates and 9-mm discs of dentine in 24-well plates (2×104 and 1×105 cells/well). After plating, all cells were incubated with 20 ng/mL rhM-CSF and 100 ng/mL receptor activator of nuclear factor-κB ligand (RANKL, PeproTech) for 11 d, with medium replacement every 3 d. Cells were subsequently fixed in paraformaldehyde for light microscopic analysis or glutaraldehyde for electron microscopic analysis. Osteoclasts were also stained for tartrate-resistant acid phosphatase (TRAP) using naphthol-ASBI-phosphate substrate18 (Sigma-Aldrich, Leukocyte Acid Phosphatase, 387A-1KT) and analyzed using an inverted microscope equipped with a CCD camera (DMI 3000B/DFC 450C, Leica). Resorption pits on fixed dentine discs were subjected to ultrasonic cleaning with distilled water at 50 kHz for 5 min, dehydrated with an ethanol gradient (40%, 70%, 90%, 95%, 100%) for 30 s and left to dry naturally. They were then stained with 1% toluidine blue at room temperature for 5 min and washed with distilled water. After observation, the discs were cleaned ultrasonically with distilled water (5 min×3 times) for scanning electron microscopy (SEM). The specimens were fixed in 2.5% glutaraldehyde in 0.1 mol/L phosphate buffer for 2 h at 4 °C, washed three times in the same buffer and post-fixed in 1% osmium tetroxide for 2 h at 4 °C. The samples were then washed 3 times in 0.1 mol/L buffer, dehydrated through graded ethanol washes (50%, 70%, 90%, 100%×3, each for 15 min), then transferred to mixtures of 100% ethanol and amyl acetate at different volume ratios (2:1, 1:2), and finished in pure amyl acetate. Slices were prepared by the CO2 critical point drying method and coated with gold before being examined in a Hitachi SU8010 SEM at 10 kV.
Results
Genetic findings
The results of the mutations are summarized in Figure 1B. Mutations of Y23, K231, G405, and C727 occur at a highly conserved position of TCIRG1 among vertebrates (Figure 1C) and were predicted to have pathogenic effects by ACMG/AMP standards and guidelines19 (Table 2).
In family 1, we detected compound heterozygous mutations: a heterozygous c.1020+1_1020+5dup in intron 9 that was inherited from the father and another mutation in exon 2, c.66delC, which resulted in a sequence change after amino acid 23 and introduced a new stop codon after 4 new amino acids (p.Tyr23ThrfsX4). The latter was a de novo mutation.
In family 2, the proband carried compound heterozygous mutations: a heterozygous c.1213G>A mutation in exon 11 inherited from his mother that resulted in a glycine-to-arginine substitution at position 405 (p.Gly405Arg) and another heterozygous mutation in exon 18, c.2181C>A, inherited from his father, which generated a stop codon at position 727 (p.Cys727X). In combination with the late younger brother's clinical features and family history, we reasonably suspected that the younger brother shared the same heterozygous mutations with the proband.
In family 3, DNA extracted from umbilical cord indicated that the deceased proband was a compound heterozygote: a heterozygous c.962delA in exon 7 inherited from his father led to a sequence change after amino acid 231, generating a new stop codon after 48 new amino acids (p.Lys231ArgfsX48); the other heterozygous mutation, c.909 C>A in exon 9, which was inherited from his mother, resulted a stop codon at position 303 (p.Tyr303X).
In family 4, molecular analysis initially focused on common genes; however, no obvious defects could be identified. We proceeded with whole-exome sequencing of Pt 4 using previously adopted methods20. Because the mode of inheritance is likely recessive in this consanguineous family, we concentrated on homozygous nonsynonymous variants in coding sequences. We preliminarily screened osteopetrosis-correlated genes (LRP5, TNFSF11, TNFRSF11A, CLCN7, OSTM1, SNX10, PLEKHM1, CA2), but there were no mutations in these genes. Therefore, we returned to the classical Wnt/β-catenin OPG/RANKL/RANK pathway and genes closely correlated with osteoclast function. We sequenced a dozen genes but had no success in finding the correct mutation. Finally, we re-visited CLCN7 and TCIRG1 and examined the 3′ UTRs of CLCN7 and TCIRG1, which may influence gene expression. Noticeably, a single nucleotide change in intron 18 of TCIRG1 (NM_006019.3, c.2236+6T>G) was annotated in the homozygous state (G/G), and other family members all presented heterozygous (T/G) for this locus. Then, RNA extracted from his whole blood was reverse transcribed into cDNA. Gel electrophoresis of the cDNA PCR products spanning exon 17 to 19 clearly presented two abnormal transcripts (Figure 3A). Next-generation paired-end sequencing generated a total of 74180 read pairs. By identifying the read pairs, we were able to confirm three major forms of transcripts. Approximately 60938 transcripts were missing exon 18 and accounted for 82.1% of transcripts, 1358 transcripts displayed a normal sequence and accounted for 1.8%, and approximately 10332 transcripts retained intron 18 and accounted for 13.9% (Figure 3B, 3C).

Gel electrophoresis of the cDNA PCR products showing the presence of two abnormal bands in Pt 4, the band 675 bp represented the transcripts which retained intron 18, 255 bp represented the transcripts which skipped exon 18, normal band was not seen; (B) Schematic representation of the relative region of the TCIRG1 gene, highlighting the position of the mutation identified in intron 18, the nucleotide change (c.2236+6T>G) found in Pt 4 is directed to lower line written in capital letter. (B) Structure of the major three types of aberrant transcripts found in Pt 4, the first one was spliced with missed exon 18, the second one was the normal transcript, while the third one retained intron 18.
Osteoclast differentiation and function of Pt 4
We analyzed the ability of osteoclasts to differentiate in vitro from monocytes isolated from the nontransplanted TCIRG1-mutated patient with relatively mild clinical symptoms, compared to an age-matched healthy male. The growth and number of osteoclasts were assessed by TRAP staining. TRAP+ multinucleated cell numbers and cell morphology between the healthy control and the patient are similar (Figure 4). Moreover, when cultured on dentine, osteoclasts from the healthy control resorbed large pits that were observed using light microscopy (toluidine blue staining) and SEM, but no absorption lacunae were detected in the patient cultures (Figure 4).

Osteoclast generation and bone resorptive activity in vitro from individual with TCIRG1-defected ARO. PBMNCs from peripheral blood mononuclear cells of Pt 4 and one age-matched male control were concurrent cultured for 9 d with rhM-CSF, then digested and seeded on dentine discs in the presence of M-CSF and RANKL for 11 d. (A) Cells (magnification: 200×) in 96-well plates stained for TRAP, showing abundant multinucleated, TRAP-positive cells in both control and Pt 4; (B, C) Lacunae on dentine slices observed by (B) light microscopy with toluidine blue staining, 100× and (C) scanning electron microscopy. Light micrograph of the control showing varied shape of resorption lacunae scatter on the dentine, SEM image indicates circular, elliptical, sausage and irregular shaped resorption pits with typical punched-out osteoclastic excavations and exposure of collagen fibres. Bone cultures of osteopetrotic PBMNCs show no sign of absorption. Scale bar, 100 μm in the bottom of the photographs. (D) TRAP-positive multinucleated osteoclast cell numbers are shown (we calculated 5 culture holes in 96-well plates, repeated 3 times), statistical difference between two group of 96-well plates were determined by the Student's t test and analyzed by a one-way ANOVA followed by Student Neuman Keuls (S-N-K) testing.
Discussion
Human ARO is a rare genetic bone disease that is characterized by generalized increased bone density and a variety of heterogeneous symptoms, including hematological and neural defects with diverse severity21. TCIRG1 is located on chromosome 11q13, encodes the osteoclast-specific 116 kD subunit of the vacuolar proton pump, contains 20 exons, and mediates H+ transport into the resorption lacunae, where a low pH is a prerequisite for the dissolution of hydroxyapatite crystals22. Generally, patients with defective TCIRG1 display life-threatening symptoms in their early lives and urgently need HSCT.
In this study, we described five patients (from four unrelated families) who suffered from osteopetrosis. We discovered seven different TCIRG1 mutations (Figure 1B), of which five mutations (c.66delC, c.1020+1_1020+5dup, c.2181C>A, c.2236+6T>G, c.692delA) were novel based on the Exome Aggregation Consortium (ExAC) database and the Human Gene Mutation Database (HGMD). Whereas Pt 1, Pt 2A, Pt 2B and Pt 3 all manifested with severe osteopetrotic bone phenotypes and typical hematological abnormalities in infancy, Pt 4, from a consanguineous family, did not display serious symptoms until high school. His disease progression was relatively slow and benign, suggesting the existence of intermediate cases. In family 1, family 2 and family 3, compound heterozygous mutations of TCIRG1 contributed to the early onset of osteopetrosis. The de novo mutation carried by Pt 1 was suspected to be the result of an alteration in a germ cell or in the fertilized egg itself. Remarkably, the affected children from family 2 and family 3 shared two point mutations, one inherited from the mother and the other from the father. The typical rickets-like nodular costochondral junctions at the ends of ribs and the widened growth plate, also termed osteopetrorickets, presented in the malignant affected pediatric patients, but not the mildly affected patient. Sobacchi et al did not detect osteopetrorickets in affected individuals with the mild form either. Osteopetrorickets was initially suspected to be a paradoxical complication of osteopetrosis resulting from the inability of osteoclasts to maintain a normal calcium-phosphorus balance in the extracellular fluid23. In this study, patients with osteopetrorickets displayed abnormal metabolism of calcium and phosphorus.
The identification of the mutation in Pt 4 was a tortuous course. Further verification in family members and transcript analysis confirmed our suspicion that two different aberrant transcripts were produced, the most abundant being the transcripts missing exon 18, while the second were transcripts that retained intron 18. We also noticed that a small number of normal transcripts were generated, which allowed a small amount of protein with partial enzyme activity to compensate for part of the osteoclast functional defect in vivo. This finding would explain the overall benign form of ARO with slow progression. Nevertheless, in vitro osteoclast function tests showed normal quantities and morphology of cells, but they were still unable to resorb bone when exposed to rhM-CSF and RANKL co-cultures, confirming that the vacuolar proton pump plays a fundamental role in the late stage of osteoclast physiology. These intrinsic defects were unable to be rescued by rhM-CSF and RANKL.
Recently, Sobacchi et al reported that an incomplete splicing defect in TCIRG1 led to benign, recessive osteopetrosis in an 8-year-old girl. This result revealed that a mild form of TCIRG1-dependent ARO does exist9. Then, the team further discovered 3 different single nucleotide changes deeply embedded in intron 15 that were predicted to impact the splicing process of the neighboring exons, which suggested that this region may be a hot spot region for mutation10. Notably, in the present study, a mutation in the splice site c.2236+6T>G has not been described previously, but a homozygous c.2236+1G>A near this location has been reported24. The affected male was born into a consanguineous Muslim family and presented with noisy breathing, prominent eyes, and bleeding spots early in life. He was diagnosed at 18 months, and he had two older affected siblings who died in infancy. Other adjacent homozygous mutations that may exert an influence on the splicing process have also been investigated6,25, but these changes led to infantile malignant osteopetrosis. Variants affecting splice sites represent a high percentage of the molecular abnormalities in the TCIRG1 gene (44.2%), among which the invariant GT/AG dinucleotide is 30.4%25. The mutations discussed here are located close to the canonical splice sites, but their functional impact generated different outcomes.
The comparisons between the malignant form in this study and the known mild form are listed in Table 3. It is noteworthy that malignant patients displayed a disorder of calcium and phosphorus metabolism, with related osteopetrorickets, but the mild phenotype did not present these characteristics. The BTMs of the malignant patients were not available, but the BTMs of the mild phenotype were normal or slightly elevated. The patients of the mild form with known mutations all had splicing defects that exerted an influence on the latter part of the TCIRG1 protein. This finding may indicate that the last 5 exons of the TCIRG1 protein are not quite as important as the N-terminal portion, or that they are partially compensated by the limited amount of normal transcripts. Interestingly, patients with this mild form with extremely dense but brittle bones and an almost vanished medullary cavity did not result in normocytic anemia or thrombocytopenia, which highlights the strong compensatory ability of the hematopoietic tissue. There are also common features between the severe and mild phenotypes, such as dental abnormalities and dense bone. However, many complex issues remain to be resolved; for example, how do the transcripts function, in what quantity, do they function alone or are they modified by other genes? How will this benign form progress in the following years, slowly, as before, or will complications, such as nerve compression, occur?
The incidence of osteopetrosis related to the Chinese population has not yet been reported due to the immense population base. However, the number of individuals who suffer from the disease cannot be ignored. HSCT, the most widely adopted treatment abroad, has been shown to be the only cure for the prevention and reversal of bone manifestations3. However, because HSCT does not improve already existing neurological sequelae26, it should be performed as soon as possible. Unfortunately, very few domestic hospitals that are capable of performing HSCT have launched therapeutic programs for patients with osteopetrosis. In 2011 and 2012, at meetings of the IEWP-EBMT (Inborn Error Working Party of the European Group for Blood and Marrow Transplantation), a survey showed that 10 patients younger than 10 months of age survived HSCT, but almost all patients experienced complications of graft rejection or autologous engraftment reconstitution, even when using cells from an HLA-haploidentical donor27. Ordinary families in China can hardly afford the cost of treatment; thus, many patients have not been properly treated or received correct genetic counseling. The combination of molecular biology and amniocentesis promotes the application of prenatal diagnosis, which facilitates early intervention for the already suffering family based on the known pathogenic mutations.
There were several limitations in our study. First, a single case of the benign form of TCIRG1-induced ARO is not sufficient to summarize all properties of this mild phenotype. Second, some serum parameters, such as BTMs, were not available. In addition, we did not perform studies on the working mechanism of this mild phenotype and more in-depth research is needed.
In conclusion, we described a relatively large sample of TCIRG1-dependent ARO, reported for the first time a mild phenotype in the Chinese population, and enriched the database of TCIRG1 mutations. Additional mechanistic studies are needed to further elucidate the biological effects of the limited amount of aberrant transcript, which spared patients from life-threatening symptoms, and this benign case may shed light on new therapeutic interventions.
References
Johnston CC Jr, Lavy N, Lord T, Vellios F, Merritt AD, Deiss WP Jr. Osteopetrosis. A clinical, genetic, metabolic, and morphologic study of the dominantly inherited, benign form. Medicine (Baltimore) 1968; 47: 149–67.
Stark Z, Savarirayan R . Osteopetrosis. Orphanet J Rare Dis 2009; 4: 5.
Sobacchi C, Schulz A, Coxon FP, Villa A, Helfrich MH . Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol 2013; 9: 522–36.
Li YP, Chen W, Liang YQ, Li E, Stashenko P . Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat Genet 1999; 23: 447–51.
Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson JP, et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet 2000; 25: 343–6.
Sobacchi C, Frattini A, Orchard P, Porras O, Tezcan I, Andolina M, et al. The mutational spectrum of human malignant autosomal recessive osteopetrosis. Hum Mol Genet 2001; 10: 1767–73.
Kornak U, Schulz A, Friedrich W, Uhlhaas S, Kremens B, Voit T, et al. Mutations in the a3 subunit of the vacuolar H+-ATPase cause infantile malignant osteopetrosis. Hum Mol Genet 2000; 9: 2059–63.
Sobacchi C, Schulz A, Coxon FP, Villa A, Helfrich MH . Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol 2013; 9: 522–36.
Sobacchi C, Pangrazio A, Lopez AG, Gomez DP, Caldana ME, Susani L, et al. As little as needed: the extraordinary case of a mild recessive osteopetrosis owing to a novel splicing hypomorphic mutation in the TCIRG1 gene. J Bone Miner Res 2014; 29: 1646–50.
Palagano E, Blair HC, Pangrazio A, Tourkova I, Strina D, Angius A, et al. Buried in the middle but guilty: intronic mutations in the TCIRG1 gene cause human autosomal recessive osteopetrosis. J Bone Miner Res 2015; 30: 1814–21.
Yu T, Yu Y, Wang J, Yin L, Zhou Y, Ying D, et al. Identification of TCIRG1 and CLCN7 gene mutations in a patient with autosomal recessive osteopetrosis. Mol Med Rep 2014; 9: 1191–6.
Yuan P, Yue Z, Sun L, Huang W, Hu B, Yang Z, et al. Novel mutation of TCIRG1 and clinical pictures of two infantile malignant osteopetrosis patients. J Bone Miner Metab 2011; 29: 251–6.
Hu WW, Zhang Z, He JW, Fu WZ, Wang C, Zhang H, et al. Establishing reference intervals for bone turnover markers in the healthy Shanghai population and the relationship with bone mineral density in postmenopausal women. Int J Endocrinol 2013; 2013: 513925.
Zhang ZL, He JW, Zhang H, Hu WW, Fu WZ, Gu JM, et al. Identification of the CLCN7 gene mutations in two Chinese families with autosomal dominant osteopetrosis (type II). J Bone Miner Metab 2009; 27: 444–51.
Li R, Li Y, Kristiansen K, Wang J . SOAP: short oligonucleotide alignment program. Bioinformatics 2008; 24: 713–4.
Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E, Jiang T, Jiang H, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet 2010; 42: 969–72.
Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZX, Pool JE, et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 2010; 329: 75–8.
van 't Hof RJ . Osteoclast formation in the mouse coculture assay. Methods Mol Med 2003; 80: 145–52.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–24.
Zhang Z, Xia W, He J, Zhang Z, Ke Y, Yue H, et al. Exome sequencing identifies SLCO2A1 mutations as a cause of primary hypertrophic osteoarthropathy. Am J Hum Genet 2012; 90: 125–32.
Balemans W, Van Wesenbeeck L, Van Hul W . A clinical and molecular overview of the human osteopetroses. Calcif Tissue Int 2005; 77: 263–74.
Vaananen HK, Zhao H, Mulari M, Halleen JM . The cell biology of osteoclast function. J Cell Sci 2000; 113: 377–81.
Kaplan FS, August CS, Fallon MD, Gannon F, Haddad JG . Osteopetrorickets. The paradox of plenty. Pathophysiology and treatment. Clin Orthop Relat Res 1993; (294): 64–78.
Phadke SR, Fischer B, Gupta N, Ranganath P, Kabra M, Kornak U . Novel mutations in Indian patients with autosomal recessive infantile malignant osteopetrosis. Indian J Med Res 2010; 131: 508–14.
Susani L, Pangrazio A, Sobacchi C, Taranta A, Mortier G, Savarirayan R, et al. TCIRG1-dependent recessive osteopetrosis: mutation analysis, functional identification of the splicing defects, and in vitro rescue by U1 snRNA. Hum Mutat 2004; 24: 225–35.
Steward CG . Neurological aspects of osteopetrosis. Neuropathol Appl Neurobiol 2003; 29: 87–97.
Schulz AS, Moshous D, Steward C, Hoenig M, Schuetz C, Sobacchi C, et al. Osteopetrosis: a heterogeneous group of diseases requiring individualized therapeutic strategies - results of the osteopetrosis registry on behalf of ESID and EBMT. Bone Marrow Transplantation 2013; 48: S22–3.
Acknowledgements
We thank all patients and family members who participated in this study.
This work is supported by grants from the National Basic Research Program of China (973 Program, 2014CB942903), National Natural Science Foundation of China (NSFC, 81370978, 8127096), The Science and Technology Commission of Shanghai Municipality (14JC1405000), and the Science and Technology Commission of Chongqing Municipality (CSTC2013jcyjC00009). Special thanks should be given to Guo-ying ZHU from the Institute of Radiation Medicine, Fudan University for cell culture guidance.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary information is available at the website of Acta Pharmacologica Sinica.
Supplementary information
Supplementary Table S1
Primer sequences of TCIRG1 (DOC 33 kb)
Rights and permissions
About this article

Cite this article
Zhang, Xy., He, Jw., Fu, Wz. et al. Novel mutations of TCIRG1 cause a malignant and mild phenotype of autosomal recessive osteopetrosis (ARO) in four Chinese families. Acta Pharmacol Sin 38, 1456–1465 (2017). https://doi.org/10.1038/aps.2017.108
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2017.108
Keywords
This article is cited by
-
Genetics of Osteopetrosis
Current Osteoporosis Reports (2018)
