
Abstract
Aim:
The novel anticancer compound TM208 is an EGFR tyrosine kinase inhibitor (EGFR-TKI). Since the development of resistance to EGFR-TKIs is a major challenge in their clinical usage, we investigated the profiles of resistance following continuous treatment with TM208 in human breast cancer xenograft mice, and identified the relationship between the tumor pEGFR levels and tumor growth inhibition.
Methods:
Female BALB/c nude mice were implanted with human breast cancer MCF-7 cells, and the xenograft mice received TM208 (50 or 150 mg·kg−1·d−1, ig) or vehicle for 18 d. The pharmacokinetics (PK) and pharmacodynamics (PD) of TM208 were evaluated.
Results:
The PK properties of TM208 were described by a one-compartment model with first-order absorption kinetics. Our study showed the inhibitory effects of TM208 on tumor pEGFR levels gradually reached a maximum effect, after which it became weaker over time, which was characterized by a combined tolerance/indirect response PD model with an estimated EC50 (55.9 μg/L), as well as three parameters ('a' of 27.2%, 'b' of 2730%, 'c' of 0.58 h−1) denoting the maximum, extent and rate of resistance, respectively. The relationship between the tumor pEGFR levels and tumor growth inhibition was characterized by a combined logistic tumor growth/transit compartment model with estimated parameters associated with tumor growth characteristics kng (0.282 day−1), drug potency kTM208 (0.0499 cm3/day) and the kinetics of tumor cell death k1 (0.141 day−1), which provided insight into drug mechanisms and behaviors.
Conclusion:
The proposed PK/PD model provides a better understanding of the pharmacological properties of TM208 in the treatment of breast cancer. Furthermore, simulation based on a tolerance model allows prediction of the occurrence of resistance.
Similar content being viewed by others

Introduction
Epidermal growth factor receptor (EGFR) is a member of the ErbB receptor family of tyrosine kinases shown to be mechanistically involved in cell proliferation and survival. It is overexpressed in a variety of human solid tumors; this overexpression always correlates with a poor prognosis1,2,3,4,5. Furthermore, some studies have demonstrated that EGFR signaling is implicated in the self renewal of cancer cells in mammospheres6, which suggests that EGFR inhibitors may be used to eliminate breast cancer stem cells7. Therefore, the blockade of EGFR has been validated as an effective approach for cancer therapy. Unfortunately, EGFR-tyrosine kinase inhibitor (EGFR-TKI) therapy has been impeded by resistance8,9. Thus, it is important to explore effective means of characterizing patterns of resistance over time and to optimize treatment to prevent or at least prolong the time to EGFR-TKI resistance.
Pharmacokinetic/pharmacodynamic (PK/PD) models are powerful tools in drug research and development that can be applied to quantitatively characterize and predict the behaviors and actions of drugs10. In current pharmaceutical research, particularly oncology investigation and anticancer drug development, a number of validated biomarkers are commonly used as efficacy endpoints to establish PD models to clarify the relationship between drug exposure and its combined effects with PK models11,12. Several models of EGFR as an important PD biomarker have been developed13,14, but few describe the time course of EGFR levels after continuous treatment and the EGFR-TKI resistance phenomenon.
Dithiocarbamates have shown promising efficacy in preventing or treating a variety of cancers15,16; as such, a series of new dithiocarbamate derivatives, including TM208, have been synthesized (Figure 1)17,18,19. As a potent anticancer candidate, TM208 exhibited favorable anti-tumor effects with low toxicity20,21,22. Our previous studies indicated that the inhibition of phosphorylated EGFR (pEGFR) contributes to the efficacy of TM208, which prevented EGFR autophosphorylation and subsequently blocked a series of downstream signaling pathways, leading to the suppression of tumor cell proliferation and angiogenesis20. However, the inhibitory effect on pEGFR was found to decrease gradually after continuous treatment with TM208, possibly due to resistance.

The chemical structures of TM208.
The purpose of this study was to develop mathematical models to characterize the PK properties of TM208, to assess the relationship between pEGFR levels and tumor growth and to quantitatively describe the time course of resistance after repeated dosing. This study may benefit the further development of TM208 as a new anticancer compound. In addition, these models will add to our understanding of the process of EGFR-TKI resistance and provide insight into the relevant mechanisms underlying resistance.
Materials and methods
Drugs and reagents
TM208 (>99.5%) was provided by Prof Run-tao LI (Peking University)23. RPMI-1640 medium was obtained from Macgene Biotech Co, Ltd (Beijing, China). Fetal bovine serum was purchased from Gibco (Grand Island, NY, USA).
Cell culture
The MCF-7 cell line was purchased from the Institute of Materia Medica, Academy of Medical Sciences (Beijing, China). MCF-7 cells were cultured in RPMI-1640 medium, which was supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin. The cells were maintained at 37 °C in an atmosphere containing 5% CO2.
Animals
The Beijing Vital Laboratory Animal Technology (Beijing, China) provided female BALB/c nude mice (5–6 weeks old). All animal studies were approved by the Institutional Animal Care and Use Committee of Peking University Health Science Center (Beijing, China), and the experiments were conducted according to the guidelines set by the National Research Council.
Tumor xenograft model
A total of 2×106 MCF-7 cells were suspended in 200 μL PBS and then inoculated subcutaneously in both second mammary fat pads of nude mice. The tumor diameter was measured with vernier calipers and converted to tumor volume using the formula 1/2×length×width2.
Pharmacokinetic Study
A liquid chromatography–tandem mass spectrometry (LC-MS/MS) method was developed by our group to determine the pharmacokinetics of TM208 in plasma24. According to the study, TM208 was dosed by gavage at 150 mg/kg per day, and plasma samples were collected at 0, 0.5, 1, 4, 10, 24, 36, 48, 120, 192, 264, 336, 408, 409, 418, and 432 h; three mice were assessed at each time point.
Tumor growth inhibition assay
Xenograft mice were randomly divided into three groups of four mice each. TM208 was dissolved in 15% hydroxypropyl-β-cyclodextrin aqueous solution and administered by intragastric gavage at a dose of 50 or 150 mg/kg per day. The vehicle control group received only vehicle solution. Tumor size was measured every three days. After 18 d of treatment, animals were euthanized by cervical displacement. Tumors were collected after the final treatment and frozen at −80 °C until use.
Determination of pEGFR levels
Xenograft mice were randomly divided into two groups, the vehicle control group and the TM208 treatment group. Nude mice were treated with vehicle solution or TM208 (150 mg/kg per day). Tumors were collected at 0, 3, 6, 9, 12, 15, and 18 d from 3 mice at each time point. In the treatment group, tumors were also collected at 0, 1, 10, and 24 h after single dosing. In addition, healthy breast tissues were collected at 0, 9, and 18 d from 3 mice at each time point. The tumors and healthy breast tissues were washed in ice-cold saline and snap-frozen. All tumors and breast tissues were cut into small pieces, homogenized, and then sonicated for 25 s in ice-cold PBS (pH=7.4). The lysates were centrifuged at 12 000×g for 30 min at 4 °C, and the supernatants were collected. The concentration of pEGFR in 100 μg of total protein was determined using a human pEGFR ELISA kit (Beijing Puli Zhicheng Biotechnology Co, Ltd, Beijing, China).
PK/PD models
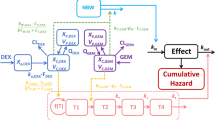
In our previous studies, the inhibition of EGFR autophosphorylation was found to be related to the antitumor properties of TM208. Furthermore, pEGFR-TKI resistance following continuous treatment was observed as time went on. In this study, a combined tolerance/indirect response model and a combined logistic tumor growth/transit compartment model were used to describe the relationship between pEGFR levels and tumor volumes. The structure of the final model is shown in Figure 2.

Schematic diagram of integrated PK/PD model that integrates the models for describing pEGFR inhibitory effects and EGFR-TKI resistance. Xa and Xc represent the absorption and central compartments, respectively; CL represents the clearance rate. kin and kout represent the rate constants for pEGFR production and output, respectively; TOLE represents the resistance induced by continuous treatment with TM208. EpEGFR is the pEGFR inhibition index, kTM208 is the antitumor effect of TM208 via pEGFR inhibition, X1 and X2-X4 represent the tumor cell proliferating compartment and non-proliferating compartments, respectively, and k1 is the transit-rate constant for the non-proliferating compartments. This model is described by equations 1–15.
PK model
The pharmacokinetic properties of TM208 were described by a one-compartment model with first-order extravascular absorption kinetics (equations 1 and 2),


in which ka is the absorption rate constant, k is the clearance rate constant and Xa and Xc represent the absorption and central compartments, respectively.
pEGFR model
Because of disease progression, the pEGFR levels varied over time both in the treatment and control tumor-bearing groups. Therefore, the pEGFR was defined as follows:

CpEGFR(0) and CpEGFR(t) represent the mean pEGFR levels at times 0 and t, respectively. All pEGFR levels at different time points were normalized to their initial levels25.
The relationship between pEGFR levels and the plasma concentration of TM208 was described using an indirect response model (equation 4),

in which kin represents the zero order rate constant of the autophosphorylation of EGFR, and kout is the first-order rate constant of the dephosphorylation process of pEGFR. Physiology is generally considered to be invariable over time at baseline; therefore the ratio of kin to kout is assumed to be constant and can be expressed as kin/kout=100%. In the model, the following equations were used to determine the effects of vehicle control (Eplacebo) and TM208 (ETM208) on pEGFR levels.

In equation 5, Pmax represents the maximum effect of vehicle control, and kplacebo is the variation rate constant of effect. The relationship between pEGFR inhibition and the plasma concentration of TM208 was described by equations 6 and 7,


in which the drug-specific parameters Emax and EC50 represent the maximum effect of pEGFR inhibition and the TM208 concentration causing one-half of the maximum effect, respectively. Emax was fixed at 1, which means pEGFR can be completely inhibited by TM208.
Tolerance model
Our previous studies showed that after continuous treatment, pEGFR levels did not continue decreasing or stabilize at a plateau; instead, they decreased in the early phase and then increased gradually as time went on, which was likely related to EGFR-TKI resistance. Therefore, a tolerance model was established to describe this resistance26,27.

The parameters a, b and c denote the maximum, development extent and development rate of EGFR-TKI resistance, respectively. A combination of the indirect response model and the tolerance model was used to describe the effect of TM208 on pEGFR levels over time.
Tumor volume model
A logistic tumor growth model was used as the base model for tumor growth dynamics (equation 9). The natural growth of the tumor can be described by an exponential phase followed by a linear growth phase, eventually reaching a plateau28,

in which kng is the first-order net growth rate constant, and Xss represents the maximum sustainable tumor volume, which is assumed to be constant.
Because the inhibition of EGFR autophosphorylation plays an important role in the antitumor efficacy of TM208, it was assumed that tumor growth might be quantitatively related to pEGFR levels. Therefore, the pEGFR profiles were used to link the final PD endpoint (tumor volume) to establish the PK/PD model.


Here, a pEGFR “inhibition index” EpEGFR (shown in equation 11) was used as a variable to describe the pEGFR inhibition28. In the TM208 treatment group, it was assumed that partial proliferating tumor cells become non-proliferating and eventually die following administration. X1 represents the portion of proliferating cells within the total tumor volume, while X2, X3 and X4 are the volumes of tumor cells at different non-proliferating stages and vt represents the sum of the volumes of tumor cells at various stages; this contains the volumes of proliferating and non-proliferating cells (equation 12). Prior to drug administration, all tumor cells are proliferating and thus X1(0)=v0 (the initial tumor volume). The time from proliferating (X1) to non-proliferating cells (X2, X3, X4) is denoted as kTM208, which describes the anti-tumor potency of TM208.
To characterize the delay in the PD response, a transit compartment model was used29,30,31. Here k1 represents the transit rate constant between any two of the non-proliferating compartments. The differential equations are shown below (equations 12–15).




Data analysis and model simulations
The model estimations were performed using NONMEM 7 (level 2.0) with the first-order conditional estimation (FOCE) method. Both interindividual and residual variability were added to the models. We assumed that the parameters and residual error variability followed a logarithmic normal distribution and that the residual error model was a mixed error model. Model selection was based on the NONMEM objective function value (OFV), parameter estimates, relative standard errors (RSE) in the estimate, and exploratory analysis of the goodness-of-fit plots. The ability of the model to describe the observed data was evaluated by a visual predictive check (VPC) of the prediction with 1000 simulations using PsN (version 3.4.2).
Using the model parameters we obtained, the tumor pEGFR level-time profiles of different groups (healthy group, vehicle-control group and TM208 treatment group) were simulated from d 0 to d 27.
Results
PK/PD model of pEGFR levels
The PK parameters of TM208 are summarized in Table 1. All parameters were estimated with good precision (RSE<30%). These results suggest that TM208 was eliminated rapidly in vivo after dosing.
The observed and simulated pEGFR level-time profiles of different groups are shown in Figure 3. The circles and crosses represent the measured and fitted pEGFR levels, respectively. After the first dose of TM208 (150 mg/kg per day), the inhibitory effect peaked at approximately 10 h and then weakened through d 1, but the pEGFR levels at 24 h were lower than the initial level. One day later, the pEGFR levels decreased with time after continuous administration and reached a minimum at d 9, after which it increased slowly due to resistance (Figure 3B). In contrast, the pEGFR of the vehicle control group increased gradually until d 9 and then remained at a high level. In the breast tissues of healthy mice, the pEGFR levels were almost invariable (Figure 3A).

(A) Observed and simulated pEGFR levels in the breast tissues of healthy nude mice and the tumors of tumor-bearing nude mice after vehicle treatment. (B) Observed and simulated pEGFR levels in the tumors of nude mice following oral administration of TM208 at 150 mg/kg per day. DV: observed values; SIM: simulated values.
A tolerance model was established to describe the resistance. We assumed that both the drug's action and resistance contributed to the regulation of the pEGFR levels and thus a combined tolerance/indirect response model was used to characterize tumor pEGFR. The VPC results indicated that the model manifested acceptable predictability, and most observed pEGFR levels fell within the 80% prediction intervals (PIs, Figure 4A–4C). The model estimates are displayed in Table 2.

(A–C) Observed and predicted pEGFR-time profiles of different groups: (A) vehicle control group; (B) healthy mice group; (C) TM208 (150 mg/kg) treatment group. (D–F) Observed and predicted tumor size-time profiles of different dose groups: (D) vehicle control group; (E) 50 mg/kg TM208 group; (F) 150 mg/kg TM208 group. The range between the dashed lines depicts the 80th percentile intervals. The solid lines represent the medians of simulated data. Solid dots represent the observed data.
The diagnostic goodness-of-fit plots for the pEGFR models are shown in Figure 5A–5D. The observed values versus either the population or the individual predicted values closely distribute around the x=y line (Figure 5A and 5B). The conditional weighted residuals (CWRES) were randomly and homogenously distributed near 0 (Figure 5C and 5D). Therefore, the models adequately predicted the observed pEGFR levels.

The goodness-of-fit plots of pEGFR model. (A) Relationship between observed versus predicted values of pEGFR; (B) Relationship between observed versus predicted individual values of pEGFR; (C) CWRES at different time points; (D) CWRES at different predicted values. The goodness-of-fit plots of the integrated model. (E) Relationship between observed versus predicted values of tumor volume; (F) Relationship between observed versus predicted individual values of tumor volume; (G) CWRES at different time points; (H) CWRES at different predicted values. CWRES: conditional weighted residuals; PRED: predicted value; IPRED: individual predicted value. The solid lines represent the x=y lines. The dotted lines are the trend lines.
PK/PD model based on tumor volume
An integrated model, which included a logistic model and a transit compartment model, was applied to describe the time course of tumor growth inhibition. Figure 4D-4F show the fitted tumor volume-time profiles of the vehicle control group (Figure 4D) and TM208 treatment groups (Figure 4E, 50 mg/kg; 4F, 150 mg/kg). The results suggest that the models fit well with the observed values, and most observed values were within the range of an 80% confidence interval of predictions. The parameter estimates are listed in Table 3. The goodness-of-fit plots, as shown in Figure 5E and 5F, indicate that the model demonstrated acceptable predictability.
Model simulation
The simulated pEGFR-time profiles of different groups (vehicle control group, TM208 treatment group and healthy mice group) dosed from d 0 to d 27 are shown in Figure 6. In the vehicle control group, the pEGFR levels increased gradually and plateaued on d 9. In the TM208 treatment group, the pEGFR levels decreased with time until d 9. Due to resistance, the pEGFR then increased slowly and eventually approached the level of normal mammary tissues; these were still much lower than those in the vehicle control group.

Simulated pEGFR-time profiles of healthy (+) and tumor-bearing (○) nude mice after vehicle treatment and multiple doses of 150 mg/kg per day TM208 (Δ) from d 0 to d 27.
Discussion
Our previous studies demonstrated that TM208 is a potential anticancer candidate that inhibits breast tumor cell proliferation both in vitro and in vivo by blocking the phosphorylation of EGFR and its downstream signaling pathway20. We observed a delay in the inhibitory effect of TM208 on pEGFR after a single dose following exposure to TM208, which indicated the duration of time involved in the transduction of the tyrosine kinases (TKs)32. To quantitatively investigate the relationship between the plasma concentration of TM208 and tumor pEGFR levels, an indirect response PD model was developed. This model describes the delayed response more adequately than the effect compartment model28,33. In this model, we assumed that TM208 affects pEGFR levels through an indirect PD effect, diminishing the formation rate of pEGFR, ie, parameter kin (1.9 h−1). In this model, we not only evaluated the effects of TM208 (ETM208), but also evaluated the placebo effect (Eplacebo) in the vehicle control group, which provided a more reasonable fit for the PK/PD relationship between drug exposure and the change in pEGFR34,35. The estimate of EC50 was 55.9 μg/L, which suggests a potent inhibitory effect of TM208 on pEGFR. Pmax indicated that the placebo effect was minimal, as the estimate was only 0.0271%.
Consistent with the observed data, the fitted pEGFR levels remained steady in the breast tissue of the healthy nude mice. However, the levels increased and subsequently reached a plateau in the vehicle control group due to status of the breast tumors36. This study showed that pEGFR in the TM208 treatment group did not continue to decrease or remain at a high level after continuous administration; instead, the inhibition of TM208 on tumor pEGFR levels gradually reached a maximum effect, after which it became weaker over time. This phenomenon was likely a result of resistance to EGFR-TKI. Therefore, a tolerance model was introduced to characterize the trajectory of resistance. Consequently, three parameters (a, b and c in Equation 8) were employed to quantify the maximum, extent and rate of resistance, respectively. As the results of VPC (Figure 4A–4C) and goodness-of-fit (Figure 5A–5D) suggested, this joint tolerance/indirect response model adequately captured the profiles of both the drug effect (inhibitory effect) and tumor resistance. To the best of our knowledge, this is the first mathematical model that describes EGFR-TKI resistance.
As the final PD endpoint, tumor volume is thought to be quantitatively related to pEGFR levels. In this study, a logistic tumor growth model was initially proposed to describe the time course of tumor growth; later this was further extended into a linked PK/PD model by incorporating the observed pEGFR levels. We considered several models to fit the natural tumor growth, including the Gompertz model, Koch model and logistic model37,38. Of these, the logistic model best fit the observations, suggesting that the time course of tumor growth after multiple dose administration could be adequately described with only two parameters, the growth rate (0.282 cm3/day) and maximum size (2.62 cm3) of the breast cancer xenografts. Among the model parameters that were identified, kTM208 is an important index of drug action being directly linked to the inhibition of tumor growth. TM208 demonstrated a similar efficacy (kTM208 of 0.349 cm3/week) to the published estimates of the antitumor effects of other EGFR inhibitors, such as erlotinib (0.507 cm3/week)13.
The transit compartment model has often been used to characterize the delayed effects of pharmacodynamic responses in the signal transduction process10,39. In this study, the model was used to describe the delay in PD, which is driven by drug exposure. To characterize the relationship between pEGFR suppression and the inhibition of tumor growth, we established a model combining a logistic model and a transit compartment model. In this model, the estimated parameters are associated with tumor growth characteristics (kng of 0.282 day−1), drug potency (kTM208 of 0.0499 cm3/day), and the kinetics of tumor cell death (k1 of 0.141 day−1). These parameters provide insight into drug mechanisms and behaviors.
Due to the limitation of sample size, it was difficult to obtain more pharmacokinetic and pharmacodynamic data to further confirm the new model; therefore, further PK and PD studies using multiple doses will be necessary.
In summary, the proposed PK/PD model provided a thorough interpretation of the relationship between the pharmacokinetic character of TM208 and its therapeutic responses, as well as the role of pEGFR inhibition in breast cancer treatment. Furthermore, the tolerance model that was established and the model-based simulation may further our understanding of the EGFR-TKI resistance process and its relevant mechanisms and eventually may contribute to avoiding resistance and maximizing therapeutic efficacy.
Author contribution
Xi-wei JI, Tian-yan ZHOU and Wei LU designed the research; Xi-wei JI, Shuang-min JI, Run-tao LI and Ke-hua WU performed the research; Xi-wei JI, Shuang-min JI, Ke-hua WU and Xiao ZHU analyzed the data; Xi-wei JI and Shuang-min JI wrote the paper.
References
Woodburn JR . The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol Ther 1999; 82: 241–50.
Nicholson RI, Gee JM, Harper ME . EGFR and cancer prognosis. Eur J Cancer 2001; 37: S9–15.
Abd El-Rehim DM, Pinder SE, Paish CE, Bell JA, Rampaul RS, Blamey RW, et al. Expression and co-expression of the members of the epidermal growth factor receptor (EGFR) family in invasive breast carcinoma. Br J Cancer 2004; 91: 1532–42.
Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW . Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res 2003; 284: 31–53.
Toi M, Osaki A, Yamada H, Toge T . Epidermal growth factor receptor expression as a prognostic indicator in breast cancer. Eur J Cancer 1991; 27: 977–80.
Farnie G, Clarke RB, Spence K, Pinnock N, Brennan K, Anderson NG, et al. Novel cell culture technique for primary ductal carcinoma in situ: role of Notch and epidermal growth factor receptor signaling pathways. J Natl Cancer Inst 2007; 99: 616–27.
Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 2008; 100: 672–9.
Mueller KL, Yang ZQ, Haddad R, Ethier SP, Boerner JL . EGFR/Met association regulates EGFR TKI resistance in breast cancer. J Mol Signal 2010; 5: 8.
Lee SY, Meier R, Furuta S, Lenburg ME, Kenny PA, Xu R, et al. FAM83A confers EGFR-TKI resistance in breast cancer cells and in mice. J Clin Invest 2012; 122: 3211–20.
Jiang W, Kim S, Zhang X, Lionberger RA, Davit BM, Conner DP, et al. The role of predictive biopharmaceutical modeling and simulation in drug development and regulatory evaluation. Int J Pharm 2011; 418: 151–60.
Jackson RC . Pharmacodynamic modelling of biomarker data in oncology. ISRN Pharmacol 2012; 2012: 590626.
Bender BC, Schindler E, Friberg LE . Population pharmacokinetic-pharmacodynamic modelling in oncology: a tool for predicting clinical response. Br J Clin Pharmacol 2015; 79: 56–71.
Wu Q, Li MY, Li HQ, Deng CH, Li L, Zhou TY, et al. Pharmacokinetic-pharmacodynamic modeling of the anticancer effect of erlotinib in a human non-small cell lung cancer xenograft mouse model. Acta Pharmacol Sin 2013; 34: 1427–36.
Wang S, Guo P, Wang X, Zhou Q, Gallo JM . Preclinical pharmacokinetic/pharmacodynamic models of gefitinib and the design of equivalent dosing regimens in EGFR wild-type and mutant tumor models. Mol Cancer Ther 2008; 7: 407–17.
Ge ZM, Li RT, Cheng TM, Cui JR . Synthesis and biological activities of diquaternary dipiperazinium salts containing dithiocarboxyl groups. Arch Pharm (Weinheim) 2001; 334: 173–6.
Guo BG, Ge ZM, Cheng TM, Li RT . Synthesis and anti-tumor activities of 1,4-bis[3-(amino-dithiocarboxy)propionyl] piperazine derivatives. Yao Xue Xue Bao 2001; 36: 185–7.
Li RT, Cheng TM, Cui JR . Study on the synthesis and anticancer activity of dithiocarbamate. US Patent: 1328999, 2002–01.
Li RT, Cheng TM, Cui JR . Piperazine mono(dithio)-carbamate ester compounds and analog: preparation method and pharmaceutical use. US Patent: 10/157733, 2002-05; WO Apply No: PCT/US02/16772 2002-05 2002–05.
Li RD, Zhang X, Li QY, Ge ZM, Li RT . Novel EGFR inhibitors prepared by combination of dithiocarbamic acid esters and 4-anilinoquinazolines. Bioorg Med Chem Lett 2011; 21: 3637–40.
Ji XW, Li RT, Li ZQ, Li L, Shao XY, Wang SY, et al. Inhibition of EGFR autophosphorylation plays an important role in the anti-breast cancer efficacy of the dithiocarbamate derivative TM208. Acta Pharmacol Sin 2014; 35: 239–47.
Guo W, Ran FX, Wang RQ, Cui JR, Li RT, Cheng TM, et al. Antitumour effect of hydrochloride 4-methyl-piperazine-1-carbodithioc acid 3-cyano-3,3-diphenyl-propyl ester. Chin J Clin Pharmacol Ther 2004; 9: 59–62.
Zhang N, Guo W, Wang L, Huang W, Xu B, Ge Z, et al. Effect of TM208 on QGY-7703 xenograft tumor growth. Anticancer Drugs 2008; 19: 593–8.
Jiang X, Ling X, Han F, Li R, Cui J . Studies on the metabolism of 4-methyl-piperazine-1-carbodithioc acid 3-cyano-3,3-diphenylpropyl ester hydrochloride in rats by high-performance liquid chromatography/electrospray ionization tandem mass spectrometry. J Pharm Biomed Anal 2007; 44: 1127–32.
Ji X, Chen Y, Li R, Zhou T, Lu W . A high-sensitivity LC-MS/MS method for the determination of 4-methyl-piperazine-1-carbodithioc acid 3-cyano-3,3-diphenylpropyl ester hydrochloride in rat plasma and its application to a pharmacokinetics study. Biomed Chromatogr 2012; 26: 1196–201.
Xu L, Eiseman JL, Egorin MJ, D'Argenio DZ . Physiologically-based pharmacokinetics and molecular pharmacodynamics of 17-(allylamino)-17-demethoxygeldanamycin and its active metabolite in tumor-bearing mice. J Pharmacokinet Pharmacodyn 2003; 30: 185–219.
Porchet HC, Benowitz NL, Sheiner LB . Pharmacodynamic model of tolerance: application to nicotine. J Pharmacol Exp Ther 1988; 244: 231–6.
Mandema JW, Wada DR . Pharmacodynamic model for acute tolerance development to the electroencephalographic effects of alfentanil in the rat. J Pharmacol Exp Ther 1995; 275: 1185–94.
Yamazaki S, Nguyen L, Vekich S, Shen Z, Yin MJ, Mehta PP, et al. Pharmacokinetic-pharmacodynamic modeling of biomarker response and tumor growth inhibition to an orally available heat shock protein 90 inhibitor in a human tumor xenograft mouse model. J Pharmacol Exp Ther 2011; 338: 964–73.
Mager DE, Jusko WJ . Pharmacodynamic modeling of time-dependent transduction systems. Clin Pharmacol Ther 2001; 70: 210–6.
Yang J, Mager DE, Straubinger RM . Comparison of two pharmacodynamic transduction models for the analysis of tumor therapeutic responses in model systems. AAPS J 2010; 12: 1–10.
Bernard A, Kimko H, Mital D, Poggesi I . Mathematical modeling of tumor growth and tumor growth inhibition in oncology drug development. Expert Opin Drug Metab Toxicol 2012; 8: 1057–69.
Liebmann C . Regulation of MAP kinase activity by peptide receptor signalling pathway: paradigms of multiplicity. Cell Signal 2001; 13: 777–85.
Yamazaki S, Skaptason J, Romero D, Lee JH, Zou HY, Christensen JG, et al. Pharmacokinetic-pharmacodynamic modeling of biomarker response and tumor growth inhibition to an orally available cMet kinase inhibitor in human tumor xenograft mouse models. Drug Metab Dispos 2008; 36: 1267–74.
Wang X, Shang D, Ribbing J, Ren Y, Deng C, Zhou T, et al. Placebo effect model in asthma clinical studies: longitudinal meta-analysis of forced expiratory volume in 1 second. Eur J Clin Pharmacol 2012; 68: 1157–66.
Sun W, Laughren TP, Zhu H, Hochhaus G, Wang Y . Development of a placebo effect model combined with a dropout model for bipolar disorder. J Pharmacokinet Pharmacodyn 2013; 40: 359–68.
Magkou C, Nakopoulou L, Zoubouli C, Karali K, Theohari I, Bakarakos P, et al. Expression of the epidermal growth factor receptor (EGFR) and the phosphorylated EGFR in invasive breast carcinomas. Breast Cancer Res 2008; 10: R49.
Norton L, Simon R . Growth curve of an experimental solid tumor following radiotherapy. J Natl Cancer Inst 1977; 58: 1735–41.
Koch G, Walz A, Lahu G, Schropp J . Modeling of tumor growth and anticancer effects of combination therapy. J Pharmacokinet Pharmacodyn 2009; 36: 179–97.
Hamberg AK, Dahl ML, Barban M, Scordo MG, Wadelius M, Pengo V, et al. A PK-PD model for predicting the impact of age, CYP2C9, and VKORC1 genotype on individualization of warfarin therapy. Clin Pharmacol Ther 2007; 81: 529–38.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (Grant 81273583) and the project sponsored by the research foundation of Peking University First Hospital.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Ji, Xw., Ji, Sm., Li, Rt. et al. Pharmacokinetic-pharmacodynamic modeling of the antitumor effect of TM208 and EGFR-TKI resistance in human breast cancer xenograft mice. Acta Pharmacol Sin 37, 825–833 (2016). https://doi.org/10.1038/aps.2016.40
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2016.40
Keywords
This article is cited by
-
Psoralen induced cell cycle arrest by modulating Wnt/β-catenin pathway in breast cancer cells
Scientific Reports (2018)
