
Abstract
Farnesoid X receptor (FXR) is a member of the nuclear receptor family and a ligand-modulated transcription factor. In the liver, FXR has been considered a multi-functional cell protector and a tumor suppressor. FXR can suppress liver carcinogenesis via different mechanisms: 1) FXR maintains the normal liver metabolism of bile acids, glucose and lipids; 2) FXR promotes liver regeneration and repair after injury; 3) FXR protects liver cells from death and enhances cell survival; 4) FXR suppresses hepatic inflammation, thereby preventing inflammatory damage; and 5) FXR can directly increase the expression of some tumor-suppressor genes and repress the transcription of several oncogenes. However, inflammation and epigenetic silencing are known to decrease FXR expression during tumorigenesis. The reactivation of FXR function in the liver may be a potential therapeutic approach for patients with liver cancer.
Similar content being viewed by others

Introduction
The farnesoid X receptor (FXR) is a ligand-modulated transcription factor and a member of the nuclear receptor family. FXR was originally cloned by Seol et al1 and Forman et al2 in 1995, and the subsequent reports from three labs revealed that bile acids (BAs) were endogenous agonists of FXR3,4,5. In the following years, FXR was found not only to participate in the regulation of the BA levels6,7 and lipid and glucose metabolism8, but also play important roles in regulating liver regeneration9, hepatic fibrosis10,11, cholestasis12, hepatic inflammation13,14, and immune responses15,16,17,18. Therefore, FXR is a multi-functional cell protector in the liver.
In 2007, FXR knockout (FXR−/−) mice were found to spontaneously develop liver tumors when they aged19,20. Interest in the potential function of FXR in cancer surged thereafter. Compared with matched normal tissues, FXR expression is significantly reduced in human tumor specimens21,22,23,24,25, and the downregulation of FXR is associated with malignant clinicopathological characteristics23,26. Loss of FXR leads to early mortality and/or promotes intestinal carcinogenesis in the mice with either a mutated APC gene (APCmin/+) or chronic colitis26,27,28. FXR deficiency also facilitates the progression of colorectal adenocarcinoma in C57BL/6 mice treated with the colon carcinogen azoxymethane28. A gain-of-function study using a xenograft mouse model showed that overexpression of FXR or treatment with FXR agonists represses cancer cell proliferation in vitro and xenograft growth in nude mice in vivo23,27,29. Interestingly, long-lived little mice have high levels of FXR and do not develop liver cancer after treatment with the chemical carcinogen diethylnitrosamine (DEN)24. These studies strongly suggest that FXR is a tumor suppressor and acts as an intriguing bridge between metabolic regulation and tumor development. The structure of FXR and the roles of FXR in the regulation of various metabolic processes have been previously described in detail8,30,31,32,33,34; here, we will summarize the findings related to the roles of FXR in liver carcinogenesis, which remains a major burden of cancer morbidity around the world.
FXR, bile acid and liver cancer
Our understanding of the role of BAs has evolved from physiological detergents essential for lipid absorption to hormone-like signaling molecules30,31. Excessive accumulation of BAs has a cytotoxic effect and is considered an important etiology of tumorigenesis35. As an endogenous BA sensor, FXR is abundantly expressed in tissues participating in the BA enterohepatic circulation, such as the liver and lower digestive tract31. The primary roles of FXR are to maintain BA homeostasis and prevent BA-induced toxicity by eliciting transcriptional alterations of genes involved in BA synthesis, transportation, conjugation and detoxification36.
FXR tightly controls BA synthesis through the induction of the hepatic small heterodimer partner (SHP, NR0B2)6 and ileal fibroblast growth factor 15/19 (FGF15/19, mouse FGF15 or human homolog FGF19)7. In the liver, both SHP and FGF15/19 trans-repress the expression of cholesterol 7α-hydroxylase (CYP7A1), which is the rate-limiting enzyme in the classic BA synthesis pathway6.
BA synthesis is mainly under the control of the FXR and SHP axis; however, FXR is not exclusively epistatic to SHP in a linear regulatory pathway37,38,39. Mice with a combined loss of FXR and SHP demonstrate more severe liver injury and much greater BA overload than that in either single knockout mouse37. This study exhibits that the two nuclear receptors act synergistically to maintain BA homeostasis.
Disruption of BA metabolism is the major defect discovered in FXR−/− mice with spontaneous hepatocarcinogenesis19,20. The BAs in both serum and liver were elevated in these mice19. When the BA pool was lowered by 2% cholestyramine food, the malignant lesions were statistically reduced19. Overload of BAs due to the depletion of the FXR gene is the causative factor for injury of liver cells, induction of chronic inflammation, enhancement of cell proliferation, and development of liver tumors19,20,40. In FXR−/−SHP−/− double-knockout (DKO) mice, the sharply elevated BA levels lead to the activation of the Yes-associated protein (YAP)41, which is a core component of the Hippo pathway and has recently been considered a crucial promoter of hepatocarcinogenesis42,43,44. The activation of YAP by BAs is concentration dependent. The physiological concentration or the modest elevation of BA levels, such as in exclusively FXR or SHP individual knockout mice, cannot lead to YAP activation41.
Consistent with the observation that elevated BA levels in mice fed with a 0.2% cholic acid diet significantly promoted N-nitrosodiethylamine-initiated hepatocellular carcinoma (HCC)19 formation, Lozano et al45 revealed that intrahepatic BA accumulation in bile-duct-ligated rats facilitated the carcinogenic effects of thioacetamide metabolites in cholangiocarcinoma development. The persistently high levels of BA enhanced the inflammation and bile duct proliferation, and led to the downregulation of FXR expression. Those data indicate that during hepatocarcinogenesis, BAs may function as tumor promoters as well as DNA-damaging initiators19,45. The imbalanced ratio of free and conjugated BAs also promotes the growth of human cholangiocarcinoma via FXR46. Further investigations of human clinical samples are needed to reveal the role of BA-mediated FXR signaling in the tumorigenesis of the human liver.
FXR, nonalcoholic fatty liver disease (NAFLD) and liver cancer
NAFLD is a spectrum of chronic, progressive liver diseases characterized by hepatic steatosis and is closely related to obesity and metabolic syndrome47,48. NAFLD is globally prevalent now due to the obesity epidemic and has become a major public health problem because a significant portion of obese patients will progress to having nonalcoholic steatohepatitis (NASH), and no specific therapies are currently approved for NAFLD or NASH47,48,49,50. NASH individuals demonstrate serious liver injuries including hepatocyte damage, inflammation and fibrosis47,48,49,50. Moreover, epidemiological evidence has proved that NASH and its two major comorbidities, obesity and diabetes mellitus (DM), increase the risk of HCC, especially when NASH-related cirrhosis has developed47. Some metabolic or stress-response pathways including one-carbon metabolism, NF-κB, PTEN, and microRNAs may be involved in the regulation of NASH-mediated hepatocarcinogenesis47.
As a multipurpose metabolic regulator, FXR activation inhibits the transition of NAFLD to NASH via maintaining the homeostasis of glucose, lipid and energy metabolism as well as by antagonizing hepatic inflammation and fibrogenesis, two key pathological features of NASH11,50,51. Evidence from various mouse models and clinical studies have shown that FXR activation by its agonists have beneficial effects on the treatment of NASH.
Type 2 DM has been considered an independent risk factor for HCC52. Insulin resistance is a key player in the pathogenesis of type 2 DM and a main driver of NASH51. Both administration of the FXR agonist GW4064 or exogenous overexpression of FXR significantly increased insulin sensitivity and improved glucose tolerance in db/db or ob/ob mice53,54. In contrast, FXR−/− mice demonstrated insulin resistance in the liver and peripheral tissue53. FXR deficiency also increases the susceptibility to developing NASH in a low-density lipoprotein receptor-knockout mouse fed with a high-fat diet55. The FXR agonist WAY362450 has been shown to protect against NASH by reducing hepatic inflammation and fibrosis in mice fed a methionine and choline-deficient (MCD) diet56. MCD-fed mice share a similar hepatic manifestation as human NASH57. Obeticholic acid (OCA; INT-747) is a 6α-ethyl derivative of CDCA. The results from several animal models indicate that OCA treatment ameliorates hepatic steatosis, inflammation and fibrosis49. In leptin receptor mutated Zucker (fa/fa) rats, which display similarities to the clinical features of NAFLD/NASH patients58, OCA reverses insulin resistance, alleviates lipid abnormalities and reduces the severity of the liver steatosis58. At present, OCA is the first selective FXR agonist to enter phase 2 clinical trials51. OCA mediated FXR activation has been shown to enhance insulin sensitivity in patients with type 2 DM and NAFLD51. Consistent with these data, hepatic FXR expression is significantly downregulated in NAFLD patients59. Activation of FXR may be effective to retune NAFLD-related metabolic disorders and impede the progress of NAFLD-NASH-HCC.
FXR, liver regeneration/repair and liver cancer
Liver regeneration (LR) after partial hepatectomy (PH) is a complex process of compensatory hyperplasia driven by the replication of remaining hepatocytes and is regulated by a well-cooperated network of signaling pathways, such as growth factors, cytokines and transcription factors60. Huang et al found that FXR-dependent BA signaling was required for normal LR9. In response to the increased BA flux after 70% PH, FXR activates hepatic SHP and intestinal FGF15, which results in the suppression of Cyp7A1 and BA synthesis9,61,62. Another FXR target gene, the bile salt export pump (BSEP), a canalicular BA effluxer63, can also be induced to enhance BA export9. In parallel, FXR directly promotes liver regrowth by activating the proliferative transcription factor FoxM1b9. Consistent with this result, FXR is able to alleviate age-related proliferation defects by transcriptional activation of FoxM1b in the mouse regenerating livers64. However, the impaired FXR activities in SIRT1 (a histone deacetylase) transgenic mice due to the persistent deacetylation and lower protein expression of FXR result in defective hepatocyte proliferation in the regenerating liver65.
FXR deficient mice not only exhibit delayed LR after 70% PH9,66 but also demonstrate defective repair ability in the damaged liver. When FXR is knocked down, the effect of anti-apoptosis on liver cells is compromised under the condition of serum deprivation or food withdrawal67. Meng et al17 reported that FXR−/− mice suffered from more severe CCl4-induced liver damage, marked by increased hepatocyte death and intrahepatic cholestasis. CYP27−/− mice that underwent 70% PH or CCl4 treatment displayed impaired LR or liver repair capacity due to the low BA levels in these animals, which indicates that sufficient FXR activities are required for normal LR or liver repair9,68. An injured liver will be unable to complete normal regeneration if FXR is deleted, thereby resulting in repeated cycles of cell death and compensatory liver regeneration. A previous report suggests that irregular proliferation of hepatocytes is a risk factor in promoting hepatocarcinogenesis69.
Interestingly, liver transplant is an optional therapy for liver cancer patients in advanced stages and follows the same principles as those that regulate LR after PH in the laboratory animals60. Understanding the mechanisms of LR, such as the roles of BA-FXR signaling in this complicated course, is helpful for the development of new therapeutic strategies for many severe liver diseases, including cirrhosis and cancer.
FXR, hepatic inflammation and liver cancer
HCC is the most common primary liver cancer, and the incidence is rising worldwide largely due to hepatitis B virus (HBV) and hepatitis C virus (HCV) infection, alcohol abuse, and obesity-associated NASH50,70. Overall, 90% of HCC cases occur in the setting of unresolved inflammation and subsequent severe fibrosis, regardless of the etiology70.
Evidence suggests that FXR demonstrates anti-inflammatory properties in the liver. Activation of FXR protects against concanavalin A-induced autoimmune hepatitis16 and alleviates LPS-mediated hepatic inflammation13. The proinflammatory cytokines, such as IL-6, are strong inducers of the activation of signal transducer and activator of the transcription 3 (STAT3) signaling pathway71,72,73. STAT3 protein is considered an indispensable participant in fostering proliferation and resisting apoptosis of tumor cells74,75. He et al analyzed 52 human HCC clinical samples and found that approximately 60% of the specimens had increased nuclear phospho-STAT3 and that the activation of STAT3 was associated with adverse characteristics of the tumor76. In FXR−/− mice, increased BAs mediate upregulation of cytokine IL-6 and the reduction of suppressor of cytokine signaling 3 (SOCS3, a feedback inhibitor of STAT3), which collectively lead to the constitutive activation of STAT372.
Another essential contributor of hepatocarcinogenesis is the transcription factor nuclear factor-κB (NF-κB). NF-κB is a master regulator of inflammatory signaling pathway, and it can also be modulated by proinflammatory cytokines. In the liver, NF-κB provides a central link between hepatic damage, fibrosis and HCC. It is also considered a promoter of liver carcinogenesis77,78. Wang et al revealed negative crosstalk between FXR and the NF-κB signaling pathway. On the one hand, activation of FXR inhibits NF-κB transcriptional activity via decreasing the binding between NF-κB and DNA. On the other hand, FXR transactivity is antagonized by LPS-induced NF-κB activation13. As the two key players in liver inflammation and cancer, NF-κB and STAT3 cooperate to respond to various stimuli including proinflammatory cytokine IL-6 and upregulation of pro-proliferative and anti-apoptotic genes. This will drive the development of liver cancer74. Together, these data suggest that the inhibition of NF-κB and STAT3 pathways may be another possible mechanism contributing to the FXR function as a liver tumor suppressor.
FXR-regulated target genes and liver cancer
SHP is one of the most strongly induced genes by FXR. SHP is an atypical orphan nuclear receptor, as it lacks a DNA binding domain and serves as a pivotal co-repressor via inhibiting transactivation of specific nuclear receptor partners79. SHP is considered a tumor suppressor80 in addition to a metabolic regulator81. SHP null mice spontaneously develop HCC at 12 to 15 months of age82, and SHP expression is diminished in human HCC samples and cell lines23,83. SHP represses tumor growth via the inhibition of cellular proliferation82,83 and the activation of apoptotic signals84,85. He et al observed that SHP was downregulated by promoter hypermethylation, which is an important epigenetic event during HCC development83.
As mentioned above, both FXR and SHP are liver tumor inhibitors. Although the role of the FXR-SHP axis in the BA metabolism has been well documented, the interplay between FXR and SHP in liver carcinogenesis remains unclear. The level of SHP expression was much lower in aging FXR−/− mice with HCC compared to young FXR−/− mice19. In human HCC specimens, expression of FXR and SHP are both markedly decreased23,25, and the FXR mRNA level was significantly and positively correlated with the SHP mRNA level in HCC tissues23. Those studies demonstrate that loss of SHP may contribute to liver carcinogenesis in livers deficient in FXR. Although hepatocyte-specific SHP overexpression does not reduce the liver tumor incidence and size or blunt the activation of the IL-6/STAT3 signaling pathway in the FXR−/− mice, the increased SHP expression can lower the hepatocellular dysplasia, reduce the inflammatory cell infiltration and enhance apoptosis71. SHP may partially protect against HCC development in FXR null mice71. Overall, these findings imply that FXR and SHP may act coordinately to perform their liver tumor inhibitory functions, but limited information is available regarding the exact molecular mechanism of interaction between the two nuclear receptors during liver carcinogenesis.
BSEP is another FXR key target gene that is vital to keep the normal BA levels in the hepatocytes by acting as as a canalicular BA effluxer63. The absence of BSEP can cause severe cholestasis and HCC in young children86,87. However, BSEP expression is dramatically reduced in human HCC samples88. BSEP is transcriptionally induced by FXR in an isoform-specific manner88. FXRα1 and FXRα2 are two main isoforms of FXR expressed in normal human liver89, and FXRα2 has a greater ability to transactivate BSEP88. The increase in the FXRα1/FXRα2 ratio due to the stimulation of proinflammatory cytokines results in significant downregulation of BSEP88.
Another FXR targeted tumor suppressor gene is N-myc downstream-regulated gene 2 (NDRG2). NDGR2 mRNA is diminished in livers of FXR−/− mice and human HCC patients. FXR agonists or ectopic overexpression of FXR leads to the transcriptional induction of the NDRG2 gene. Furthermore, FXR can bind to the intronic IR1-type element(s) of the NDRG2 gene in mouse liver and human liver cancer cells29.
FXR also modulates the function of oncogenes. Several mouse models and clinical investigations provide direct evidence that during the development of mouse and human liver cancer, loss or reduction of FXR expression levels will cause the de-repression of the promoter of the oncoprotein gankyrin, which is a small subunit of the proteasome and mediates the degradation or elimination of activities of four tumor suppressor proteins: Rb, p53, hepatocyte nuclear factor 4a (HNF4a), and CCAAT/enhancer binding protein (C/EBP) α24.
Hepatocarcinogenesis is a multistep process with accumulation of sequential genetic and epigenetic alterations. FXR may exert its tumor inhibitory function partially via direct inactivation of oncogenes and activation of tumor-suppressor genes. More FXR target genes related to the liver carcinogenesis may be identified in the future.
Downregulation of FXR expression in liver cancer
As mentioned above, the expression of FXR is lost during the development of liver cancer. However, the precise molecular mechanism of FXR downregulation remains elusive in the current field.
Inflammation may provide a microenvironment to reduce the expression of FXR. The elevated levels of proinflammatory cytokines, such as TNFα, IL-1β, and IL-6, in both FXR−/− mice19,20 and in most human HCC patients25,88, may reduce the FXR expression via inhibiting the transactivity of hepatic nuclear factor 1α (HNF1α) on the FXR gene promoter25. TNFα and IL-1β alter the relative expression of FXRα1 and FXRα2, which leads to an increase in the FXRα1/ FXRα2 ratio and subsequent reduction of BSEP, indicting a potential interaction between FXR alternative splicing procedure and the inflammatory-cytokine mediated signal pathway88. Gadaleta et al90 demonstrated that a vicious cycle was formed when NF-κB-dependent reduction of FXR expression led to less inhibition of NF-κB-mediated intestinal inflammation (Figure 1).

A vicious cycle of FXR and liver inflammation. FXR antagonizes NF-κB activity and suppresses hepatic inflammation. Conversely, hepatic inflammation decreases FXR expression.
Epigenetic silencing is another important contributor to the reduction of FXR expression. Two studies have demonstrated a role of miR-421 in the suppression of FXR transcription by targeting the 3′UTR of FXR mRNA in HCC cells and biliary tract cancer cells91,92. However, overexpression of SIRT1 in transgenic mice results in persistent deacetylation and decreased protein expression of FXR. In human HCC, elevated SIRT1 is correlated with the absence of FXR65. Recently, Bailey et al21 reported that diminished FXR expression in human colon cancer was partly due to both DNA methylation of the FXR promoter and increased KRAS signaling. Therefore, activation of oncogenic pathways may also be responsible for the reduced expression of FXR in liver cancer.
Conclusion and perspectives
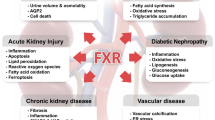
Physiologically, FXR displays its hepatoprotective roles in the following ways: 1) Maintains normal liver homeostasis and metabolism of BAs, glucose and lipid; 2) Participates in LR and acts as an important factor to reestablish BA homeostasis and promote liver regeneration; 3) Protects liver cells from further damage by inhibiting cell death; 4) Counter-regulates hepatic inflammation through suppression of NF-κB- and STAT3- mediated signaling pathways; and 5) Induces the expression of tumor-suppressor genes and represses the transcription of oncogenes (Figure 2). In addition, FXR may function in other metabolic processes, such as cholestasis and fibrosis, to suppress liver carcinogenesis, although these roles are not covered in this review.

FXR and anti-liver carcinogenesis. FXR suppresses liver carcinogenesis via several diverse mechanisms: 1) FXR maintains the normal liver homeostasis and metabolism of BAs, glucose, and lipid. 2) FXR promotes liver regeneration and repair after injury; 3) FXR suppresses hepatic inflammation; 4) FXR enhances hepatocyte cell survival; and 5) FXR increases the expression of some tumor suppressor genes and represses the transcription of several oncogenes.
Loss of FXR activity could be an important molecular event in the initiation and progression of liver cancer. Inflammation and epigenetic silencing are the two main contributors to the downregulation of FXR expression. Due to the deficiency of FXR function, hepatocytes will be exposed to a microenvironment that favors malignant transformation and cancer development. Interestingly, changing the FXR silencing or activation of remnant FXR in the healthy tissues adjacent to the tumor may be a potential therapeutic strategy for liver cancer patients.
References
Seol W, Choi HS, Moore DD . Isolation of proteins that interact specifically with the retinoid X receptor: two novel orphan receptors. Mol Endocrinol 1995; 9: 72–85.
Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995; 81: 687–93.
Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science 1999; 284: 1362–5.
Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science 1999; 284: 1365–8.
Wang H, Chen J, Hollister K, Sowers LC, Forman BM . Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell 1999; 3: 543–53.
Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell 2000; 6: 517–26.
Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2005; 2: 217–25.
Claudel T, Staels B, Kuipers F . The Farnesoid X receptor: a molecular link between bile acid and lipid and glucose metabolism. Arterioscler Thromb Vasc Biol 2005; 25: 2020–30.
Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, Liu J, et al. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science 2006; 312: 233–6.
Li J, Kuruba R, Wilson A, Gao X, Zhang Y, Li S . Inhibition of endothelin-1-mediated contraction of hepatic stellate cells by FXR ligand. PLoS One 2010; 5: e13955.
Fiorucci S, Antonelli E, Rizzo G, Renga B, Mencarelli A, Riccardi L, et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology 2004; 127: 1497–512.
Liu Y, Binz J, Numerick MJ, Dennis S, Luo G, Desai B, et al. Hepatoprotection by the farnesoid X receptor agonist GW4064 in rat models of intra- and extrahepatic cholestasis. J Clin Invest 2003; 112: 1678–87.
Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W . Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008; 48: 1632–43.
Nijmeijer RM, Gadaleta RM, van Mil SW, van Bodegraven AA, Crusius JB, Dijkstra G, et al. Farnesoid X receptor (FXR) activation and FXR genetic variation in inflammatory bowel disease. PLoS One 2011; 6: e23745.
Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S . The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol 2009; 183: 6251–61.
Mencarelli A, Renga B, Migliorati M, Cipriani S, Distrutti E, Santucci L, et al. The bile acid sensor farnesoid X receptor is a modulator of liver immunity in a rodent model of acute hepatitis. J Immunol 2009; 183: 6657–66.
Meng Z, Wang Y, Wang L, Jin W, Liu N, Pan H, et al. FXR regulates liver repair after CCl4-induced toxic injury. Mol Endocrinol 2010; 24: 886–97.
Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A 2006; 103: 3920–5.
Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W . Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res 2007; 67: 863–7.
Kim I, Morimura K, Shah Y, Yang Q, Ward JM, Gonzalez FJ . Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis 2007; 28: 940–6.
Bailey AM, Zhan L, Maru D, Shureiqi I, Pickering CR, Izzo J, et al. FXR silencing in human colon cancer by DNA methylation and KRAS signaling. Am J Physiol Gastrointest Liver Physiol 2014; 306: G48–58.
Torres J, Bao X, Iuga AC, Chen A, Harpaz N, Ullman T, et al. Farnesoid X receptor expression is decreased in colonic mucosa of patients with primary sclerosing cholangitis and colitis-associated neoplasia. Inflamm Bowel Dis 2013; 19: 275–82.
Su H, Ma C, Liu J, Li N, Gao M, Huang A, et al. Downregulation of nuclear receptor FXR is associated with multiple malignant clinicopathological characteristics in human hepatocellular carcinoma. Am J Physiol Gastrointest Liver Physiol 2012; 303: G1245–53.
Jiang Y, Iakova P, Jin J, Sullivan E, Sharin V, Hong IH, et al. Farnesoid X receptor inhibits gankyrin in mouse livers and prevents development of liver cancer. Hepatology 2013; 57: 1098–106.
Liu N, Meng Z, Lou G, Zhou W, Wang X, Zhang Y, et al. Hepatocarcinogenesis in FXR−/− mice mimics human HCC progression that operates through HNF1alpha regulation of FXR expression. Mol Endocrinol 2012; 26: 775–85.
Lax S, Schauer G, Prein K, Kapitan M, Silbert D, Berghold A, et al. Expression of the nuclear bile acid receptor/farnesoid X receptor is reduced in human colon carcinoma compared to nonneoplastic mucosa independent from site and may be associated with adverse prognosis. Int J Cancer 2012; 130: 2232–9.
Modica S, Murzilli S, Salvatore L, Schmidt DR, Moschetta A . Nuclear bile acid receptor FXR protects against intestinal tumorigenesis. Cancer Res 2008; 68: 9589–94.
Maran RR, Thomas A, Roth M, Sheng Z, Esterly N, Pinson D, et al. Farnesoid X receptor deficiency in mice leads to increased intestinal epithelial cell proliferation and tumor development. J Pharmacol Exp Ther 2009; 328: 469–77.
Deuschle U, Schuler J, Schulz A, Schluter T, Kinzel O, Abel U, et al. FXR controls the tumor suppressor NDRG2 and FXR agonists reduce liver tumor growth and metastasis in an orthotopic mouse xenograft model. PLoS One 2012; 7: e43044.
Houten SM, Watanabe M, Auwerx J . Endocrine functions of bile acids. EMBO J 2006; 25: 1419–25.
Schaap FG, Trauner M, Jansen PL . Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol 2014; 11: 55–67.
Wang YD, Chen WD, Moore DD, Huang W . FXR: a metabolic regulator and cell protector. Cell Res 2008; 18: 1087–95.
Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B . Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev 2009; 89: 147–91.
Lee FY, Lee H, Hubbert ML, Edwards PA, Zhang Y . FXR, a multipurpose nuclear receptor. Trends Biochem Sci 2006; 31: 572–80.
Bernstein C, Holubec H, Bhattacharyya AK, Nguyen H, Payne CM, Zaitlin B, et al. Carcinogenicity of deoxycholate, a secondary bile acid. Arch Toxicol 2011; 85: 863–71.
Kalaany NY, Mangelsdorf DJ . LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu Rev Physiol 2006; 68: 159–91.
Anakk S, Watanabe M, Ochsner SA, McKenna NJ, Finegold MJ, Moore DD . Combined deletion of Fxr and Shp in mice induces Cyp17a1 and results in juvenile onset cholestasis. J Clin Invest 2011; 121: 86–95.
Wang L, Lee YK, Bundman D, Han Y, Thevananther S, Kim CS, et al. Redundant pathways for negative feedback regulation of bile acid production. Dev Cell 2002; 2: 721–31.
Kerr TA, Saeki S, Schneider M, Schaefer K, Berdy S, Redder T, et al. Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Dev Cell 2002; 2: 713–20.
Wolfe A, Thomas A, Edwards G, Jaseja R, Guo GL, Apte U . Increased activation of the Wnt/beta-catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J Pharmacol Exp Ther 2011; 338: 12–21.
Anakk S, Bhosale M, Schmidt VA, Johnson RL, Finegold MJ, Moore DD . Bile acids activate YAP to promote liver carcinogenesis. Cell Rep 2013; 5: 1060–9.
Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci U S A 2010; 107: 1437–42.
Cai J, Zhang N, Zheng Y, de Wilde RF, Maitra A, Pan D . The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev 2010; 24: 2383–8.
Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 2009; 16: 425–38.
Lozano E, Sanchez-Vicente L, Monte MJ, Herraez E, Briz O, Banales JM, et al. Cocarcinogenic effects of intrahepatic bile acid accumulation in cholangiocarcinoma development. Mol Cancer Res 2014; 12: 91–100.
Dai J, Wang H, Shi Y, Dong Y, Zhang Y, Wang J . Impact of bile acids on the growth of human cholangiocarcinoma via FXR. J Hematol Oncol 2011; 4: 41.
Michelotti GA, Machado MV, Diehl AM . NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 2013; 10: 656–65.
Rozman D . From nonalcoholic Fatty liver disease to hepatocellular carcinoma: a systems understanding. Dig Dis Sci 2014; 59: 238–41.
Adorini L, Pruzanski M, Shapiro D . Farnesoid X receptor targeting to treat nonalcoholic steatohepatitis. Drug Discov Today 2012; 17: 988–97.
Fuchs M . Non-alcoholic Fatty liver disease: the bile Acid-activated farnesoid x receptor as an emerging treatment target. J Lipids 2012; 2012: 934396.
Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013; 145: 574–82.
Koh WP, Wang R, Jin A, Yu MC, Yuan JM . Diabetes mellitus and risk of hepatocellular carcinoma: findings from the Singapore Chinese Health Study. Br J Cancer 2013; 108: 1182–8.
Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci U S A 2006; 103: 1006–11.
Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk TH, Grefhorst A, Abdelkarim M, et al. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem 2006; 281: 11039–49.
Kong B, Luyendyk JP, Tawfik O, Guo GL . Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. J Pharmacol Exp Ther 2009; 328: 116–22.
Zhang S, Wang J, Liu Q, Harnish DC . Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol 2009; 51: 380–8.
Farrell GC, Larter CZ . Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 2006; 43: S99–S112.
Cipriani S, Mencarelli A, Palladino G, Fiorucci S . FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res 2010; 51: 771–84.
Yang ZX, Shen W, Sun H . Effects of nuclear receptor FXR on the regulation of liver lipid metabolism in patients with non-alcoholic fatty liver disease. Hepatol Int 2010; 4: 741–8.
Fausto N, Campbell JS, Riehle KJ . Liver regeneration. Hepatology 2006; 43: S45–53.
Zhang L, Huang X, Meng Z, Dong B, Shiah S, Moore DD, et al. Significance and mechanism of CYP7a1 gene regulation during the acute phase of liver regeneration. Mol Endocrinol 2009; 23: 137–45.
Zhang L, Wang YD, Chen WD, Wang X, Lou G, Liu N, et al. Promotion of liver regeneration/repair by farnesoid X receptor in both liver and intestine in mice. Hepatology 2012; 56: 2336–43.
Kullak-Ublick GA, Stieger B, Meier PJ . Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology 2004; 126: 322–42.
Chen WD, Wang YD, Zhang L, Shiah S, Wang M, Yang F, et al. Farnesoid X receptor alleviates age-related proliferation defects in regenerating mouse livers by activating forkhead box m1b transcription. Hepatology 2010; 51: 953–62.
Garcia-Rodriguez JL, Barbier-Torres L, Fernandez-Alvarez S, Gutierrez-de Juan V, Monte MJ, Halilbasic E, et al. SIRT1 controls liver regeneration by regulating bile acid metabolism through farnesoid X receptor and mammalian target of rapamycin signaling. Hepatology 2014; 59: 1972–83.
Borude P, Edwards G, Walesky C, Li F, Ma X, Kong B, et al. Hepatocyte-specific deletion of farnesoid X receptor delays but does not inhibit liver regeneration after partial hepatectomy in mice. Hepatology 2012; 56: 2344–52.
Wang YD, Yang F, Chen WD, Huang X, Lai L, Forman BM, et al. Farnesoid X receptor protects liver cells from apoptosis induced by serum deprivation in vitro and fasting in vivo. Mol Endocrinol 2008; 22: 1622–32.
Meng Z, Liu N, Fu X, Wang X, Wang YD, Chen WD, et al. Insufficient bile acid signaling impairs liver repair in CYP27−/− mice. J Hepatol 2011; 55: 885–95.
Ueno Y, Moriyama M, Uchida T, Arakawa Y . Irregular regeneration of hepatocytes is an important factor in the hepatocarcinogenesis of liver disease. Hepatology 2001; 33: 357–62.
Elsharkawy AM, Mann DA . Nuclear factor-kappaB and the hepatic inflammation-fibrosis-cancer axis. Hepatology 2007; 46: 590–7.
Li G, Kong B, Zhu Y, Zhan L, Williams JA, Tawfik O, et al. Small heterodimer partner overexpression partially protects against liver tumor development in farnesoid X receptor knockout mice. Toxicol Appl Pharmacol 2013; 272: 299–305.
Li G, Zhu Y, Tawfik O, Kong B, Williams JA, Zhan L, et al. Mechanisms of STAT3 activation in the liver of FXR knockout mice. Am J Physiol Gastrointest Liver Physiol 2013; 305: G829–37.
Meng Z, Wang X, Gan Y, Zhang Y, Zhou H, Ness CV, et al. Deletion of IFNgamma enhances hepatocarcinogenesis in FXR knockout mice. J Hepatol 2012; 57: 1004–12.
Grivennikov SI, Karin M . Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev 2010; 21: 11–9.
Yu H, Jove R . The STATs of cancer — new molecular targets come of age. Nat Rev Cancer 2004; 4: 97–105.
He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T, et al. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 2010; 17: 286–97.
Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004; 431: 461–6.
Luedde T, Schwabe RF . NF-kappaB in the liver — linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2011; 8: 108–18.
Seol W, Choi HS, Moore DD . An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science 1996; 272: 1336–9.
Zhang Y, Hagedorn CH, Wang L . Role of nuclear receptor SHP in metabolism and cancer. Biochim Biophys Acta 2011; 1812: 893–908.
Boulias K, Katrakili N, Bamberg K, Underhill P, Greenfield A, Talianidis I . Regulation of hepatic metabolic pathways by the orphan nuclear receptor SHP. EMBO J 2005; 24: 2624–33.
Zhang Y, Xu P, Park K, Choi Y, Moore DD, Wang L . Orphan receptor small heterodimer partner suppresses tumorigenesis by modulating cyclin D1 expression and cellular proliferation. Hepatology 2008; 48: 289–98.
He N, Park K, Zhang Y, Huang J, Lu S, Wang L . Epigenetic inhibition of nuclear receptor small heterodimer partner is associated with and regulates hepatocellular carcinoma growth. Gastroenterology 2008; 134: 793–802.
Zhang Y, Soto J, Park K, Viswanath G, Kuwada S, Abel ED, et al. Nuclear receptor SHP, a death receptor that targets mitochondria, induces apoptosis and inhibits tumor growth. Mol Cell Biol 2010; 30: 1341–56.
Zhang Y, Wang L . Nuclear receptor small heterodimer partner in apoptosis signaling and liver cancer. Cancers (Basel) 2011; 3: 198–212.
Knisely AS, Strautnieks SS, Meier Y, Stieger B, Byrne JA, Portmann BC, et al. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology 2006; 44: 478–86.
Strautnieks SS, Byrne JA, Pawlikowska L, Cebecauerova D, Rayner A, Dutton L, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology 2008; 134: 1203–14.
Chen Y, Song X, Valanejad L, Vasilenko A, More V, Qiu X, et al. Bile salt export pump is dysregulated with altered farnesoid X receptor isoform expression in patients with hepatocellular carcinoma. Hepatology 2013; 57: 1530–41.
Zhang Y, Kast-Woelbern HR, Edwards PA . Natural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activation. J Biol Chem 2003; 278: 104–10.
Gadaleta RM, Oldenburg B, Willemsen EC, Spit M, Murzilli S, Salvatore L, et al. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-kappaB signaling in the intestine. Biochim Biophys Acta 2011; 1812: 851–8.
Zhang Y, Gong W, Dai S, Huang G, Shen X, Gao M, et al. Downregulation of human farnesoid X receptor by miR-421 promotes proliferation and migration of hepatocellular carcinoma cells. Mol Cancer Res 2012; 10: 516–22.
Zhong XY, Yu JH, Zhang WG, Wang ZD, Dong Q, Tai S, et al. MicroRNA-421 functions as an oncogenic miRNA in biliary tract cancer through down-regulating farnesoid X receptor expression. Gene 2012; 493: 44–51.
Acknowledgements
This work was supported by the National Natural Science Foundation of China Grant 30972927 (Xiong-fei HUANG), Foundation of Fujian Educational Committee Grant JA09111 (Xiong-fei HUANG) and NCI 1R01-CA139158 (Wen-dong HUANG).
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Huang, Xf., Zhao, Wy. & Huang, Wd. FXR and liver carcinogenesis. Acta Pharmacol Sin 36, 37–43 (2015). https://doi.org/10.1038/aps.2014.117
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2014.117
Keywords
This article is cited by
-
The essential roles of FXR in diet and age influenced metabolic changes and liver disease development: a multi-omics study
Biomarker Research (2023)
-
Bile acids and their receptors: modulators and therapeutic targets in liver inflammation
Seminars in Immunopathology (2022)
-
Mouse models of nonalcoholic steatohepatitis and their application to new drug development
Archives of Pharmacal Research (2022)
-
Targeting Farnesoid X receptor (FXR) for developing novel therapeutics against cancer
Molecular Biomedicine (2021)
-
The role of farnesoid X receptor in metabolic diseases, and gastrointestinal and liver cancer
Nature Reviews Gastroenterology & Hepatology (2021)
