
Abstract
Aim:
Hematoporphyrin monomethyl ether (HMME), which consists of equal amounts of isomers HMME-1 and HMME-2, is a novel porphyrin-related drug for photodynamic therapy. This study was aimed to investigate the uptake transporter-mediated selective uptake of HMME into the liver and to identify the major uptake transporter isoforms involved.
Methods:
Adult SD rats were intravenously injected with a single dose of HMME (5 mg/kg) with or without rifampicin (an inhibitor of organic anion transporting polypeptides OATP1B1 and OATP1B3, 25 mg/kg). Blood samples were collected, and HMME concentrations were measured using LC-MS/MS. Rat hepatocytes, human hepatocytes and HEK293 cells expressing OATP1B1, OATP1B3, or OATP2B1 were used to investigate the uptake of HMME or individual isomers in vitro.
Results:
Co-administration of rifampicin significantly increased the exposure of HMME isomers, and decreased the AUC ratio of HMME-1 to HMME-2 from 1.98 to 1.56. The uptake of HMME-2 into human hepatocytes and the HEK293 cells expressing OATP1B1 or OATP2B1 in vitro was 2–7 times greater than that of HMME-1, whereas OATP1B3 mediated a higher HMME-1 uptake. OATP1B1 exhibited a higher affinity for HMME-2 than for HMME-1 (the Km values were 0.63 and 5.61 μmol/L, respectively), which were similar to those in human hepatocytes. By using telmisartan (a non-specific OATP inhibitor) and rifampicin, OATP2B1 was demonstrated to account for <20% of hepatic HMME uptake.
Conclusion:
OATP1B1 is the major transporter involved in the rapid hepatic uptake of HMME, and the greater uptake of HMME-2 by OATP1B1 may lead to a lower exposure of HMME-2 than HMME-1 in humans.
Similar content being viewed by others
Introduction
Hematoporphyrin monomethyl ether (HMME), which is a mixture of equal amounts of the two isomers 8-(1-methoxyethyl)-13-(1-hydroxyethyl)-3,7,12,17-tetramethyl-21H,23H-porphine-2,18-dipropanoic acid (HMME-1) and 13-(1-methoxyethyl)-8-(1-hydroxyethyl)-3,7,12,17-tetramethyl-21H,23H-porphine-2,18-dipropanoic acid (HMME-2) (Figure 1), is a new porphyrin-related drug developed by Fudan-Zhangjiang Bio-Pharmaceutical Co Ltd (Shanghai, China). It is used in photodynamic therapy for treating port wine stains and is administered by intravenous infusion. Compared with old-generation photosensitizers, such as hematoporphyrin derivatives, HMME has a simpler composition, a light avoidance period of 1 week instead of 1 month, and stronger photodynamic effects against ovarian cancer1 and hypertrophic scar fibroblasts2.

Chemical structures of HMME-1 (A) and HMME-2 (B).
After a single intravenous injection of 5 mg/kg HMME in rats, the tissue concentration of HMME was greatest in the liver3. After an intravenous administration in rats and humans, HMME was rapidly cleared from the circulation and was predominantly excreted unchanged in the bile/feces with approximately 58% of fecal recovery in rats3,4 and 43% in humans (unpublished data), which indicates the importance of the liver in the systemic elimination of HMME. The similar elimination profiles of HMME in rats and humans indicate that rats are appropriate model animals for investigating the mechanisms of hepatic elimination of HMME in humans.
A pilot study showed that the systemic exposure of HMME-1 was approximately double that of HMME-2, suggesting that HMME undergoes selective disposition in rats (unpublished data). However, HMME was metabolically stable when it was incubated with both rat liver microsomes (RLM) and human liver microsomes (HLM) in the presence of NADPH, suggesting that selective hepatic transport rather than metabolism may be responsible for its selective disposition. HMME is an amphipathic monoanion in physiological conditions (pH 7.4, blood), and the apparent permeability coefficients Papp value obtained from Caco-2 monolayer cells was approximately 3×10−8 cm/s (unpublished data), implying that the uptake of HMME into hepatocytes by passive diffusion could be limited. Thus, we speculated that active transport processes are involved in hepatic HMME uptake and that they cause the difference in the systemic exposure of each isomer in rats, which may also occur in humans.
A variety of uptake transporters expressed on the basolateral membrane of hepatocytes, such as Na+-taurocholate co-transporting polypeptide (NTCP), organic anion transporting polypeptide (rodents: Oatp; humans: OATP) 1B1, OATP1B3, OATP2B1, organic anion transporter 2 (rodents: Oat2; humans: OAT2), and organic cation transporter 1 (rodents: Oct1; humans: OCT1), are responsible for the hepatic uptake of various compounds from the systemic circulation. OATP1B1 and OATP1B3 are the predominant OATP isoforms expressed in the liver, and they are involved in the uptake of multiple endogenous and exogenous compounds, including bilirubin5, bosentan6,7, and statins8,9,10. Additionally, OATP2B1-mediated selective scutellarin uptake plays a key role in the much lower exposure of scutellarin than that of isoscutellarin in humans11.
Hence, this study was designed to investigate the transporter-mediated selective uptake of HMME isomers in the liver and identify the major transporter isoforms involved in this process. The findings may help predict the clinical drug-drug interactions (DDI) mediated by OATPs and design the clinical OATP DDI study.
Materials and methods
Materials
HMME (98% purity) was purchased from Chembest Research Laboratories Ltd (Shanghai, China). HMME-1 and HMME-2 were separated and purified from HMME using a Shimadzu LC-6AD semi-preparative liquid chromatography system (Kyoto, Japan). Deuterium-labeled HMME (d3-HMME) was synthesized in the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences (Shanghai, China). Estrone-3-sulfate (E3S), bromsulphthalein (BSP), rifampicin, telmisartan, dexamethasone, and Hanks' balanced salt buffer (HBSS, pH 7.4) were purchased from Sigma-Aldrich (St Louis, MO, USA). Dulbecco's modified Eagle's medium (DMEM), Williams' E medium, fetal bovine serum (FBS), L-glutamine, minimum essential medium nonessential amino acids (MEMNAA), penicillin G, and streptomycin were purchased from Invitrogen (Carlsbad, CA, USA). ITS+, 24-well or 48-well plates biocoated with collagen I or poly-D-lysine were purchased from BD Biosciences (San Jose, CA, USA). The BCA protein assay kit was purchased from Beyotime (Jiangsu, China). HEK293 cells individually transfected with OATP1B1, OATP1B3, and OATP2B1, and vector-control (mock-transfected cells) were established at HD Biosciences Co Ltd (Shanghai, China). Cryopreserved human hepatocytes were purchased from In Vitro Technologies (Baltimore, MD, USA; lots LTG and YJM) and from XenoTech LLC (Lenexa, KS, USA; lot HC2-25). All other solvents and regents were of either high-performance liquid chromatography (HPLC) or analytical grade.
Animal study
All rodent studies were carried out in accordance with the Guidelines for Care and Use of Laboratory Animals approved by the Shanghai Institute of Materia Medica, Chinese Academy of Sciences (Shanghai, China). The animals were fasted for 12 h with free access to water before the experiments. Male Sprague-Dawley rats weighing 250–300 g were randomly divided into two groups (n=4 for each group). In both groups, HMME was administered intravenously at a dose of 5 mg/kg. In the rifampicin-treated group, rifampicin dissolved in saline was intravenously injected into the tail vein at a dose of 25 mg/kg, 15 min before the administration of HMME. Rats receiving HMME alone were injected with an equivalent volume of saline. Blood samples were collected before the dose (0 h); 2, 5, 15, and 30 min after the dose; and 1, 2, 4, 6, and 8 h after the dose. Plasma samples were obtained by centrifugation at 11000×g for 5 min. All plasma samples were stored at −20 °C until analysis.
Cell culture
OATP1B1-, OATP1B3-, OATP2B1-, and mock-transfected HEK293 cells were maintained in DMEM supplemented with 10% FBS, 2 mmol/L L-glutamine, 100 units/mL penicillin G, 100 μg/mL streptomycin, and 1% MEMNAA at 37 °C in a humidified 5% CO2 atmosphere. BioCoat poly-D-lysine-coated 24-well plates were seeded with the cells at a density of 2×105 cells/well. Uptake studies were conducted 2 d after seeding.
Uptake studies using OATP-expressing HEK293 cells
Prior to the assay, the cells were rinsed three times with pre-warmed HBSS (37 °C) and equilibrated in HBSS for 10 min. The uptake studies were initiated by the addition of HBSS containing the test compounds, with or without the selected inhibitors, and were terminated at the designed time after removing the transport buffer. The cells were washed with ice-cold HBSS three times and lysed with deionized water (300 μL) by three freeze–thaw cycles. The protein content of the cell lysate was determined using the BCA protein assay kit. The concentrations of the test compounds were determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
Preparation of freshly isolated rat and cryopreserved human hepatocytes
Rat hepatocytes were freshly isolated from male Sprague-Dawley rats weighing 200–250 g by the two-step collagenase perfusion method as described in the literature12. Cryopreserved human hepatocytes were thawed quickly at 37 °C in a water bath and treated according to the manufacturer's instructions. Rat and human hepatocytes were finally suspended in Williams' E medium supplemented with 5% FBS, 2 mmol/L L-glutamine, 100 units/mL penicillin G, 100 μg/mL streptomycin, 0.1 μmol/L dexamethasone and 1% ITS+. The cell viability as determined by trypan blue exclusion was over 90%. Collagen I-coated 24-well plates were seeded with rat hepatocytes at a density of 3.5×105 viable cells/well, and collagen I-coated 48-well plates were seeded with human hepatocytes at a density of 1.5×105 viable cells/well. After incubated for 4 h at 37 °C under 5% CO2 and 95% humidity, the adherent hepatocytes were used for the uptake study.
Uptake of HMME into rat and human hepatocytes
The uptake studies were initiated by the addition of fresh HBSS containing HMME or its individual isomers, after the cells were rinsed twice and equilibrated in HBSS for 10 min at 37 or 4 °C. To screen the transporters involved, HMME uptake into the hepatocytes was assessed in the presence of selective inhibitors (50 μmol/L rifampicin for OATPs/Oatps, 10 μmol/L ketoprofen for OAT2/Oat2, or 10 μmol/L MPP+ for OCT1/Oct1)13,14,15,16 at 37 °C for 10 min. After the incubation for designed time at 37 or 4 °C, the uptake buffer was immediately aspirated, and the cells were quickly rinsed three times with ice-cold HBSS. The cells were lysed using 300 μL (rat hepatocytes) or 150 μL (human hepatocytes) deionized water by three freeze–thaw cycles. The protein content of the cell lysate was determined using the BCA protein assay kit. The concentrations of the test compounds were determined by LC-MS/MS.
LC-MS/MS analysis
The concentrations of HMME were determined using a Shimadzu LC-20AD HPLC system (Kyoto, Japan) and an API4000 triple quadrupole mass spectrometer (AB Sciex, Concord, Ontario, Canada) equipped with a TurboIonSpray ion source. Chromatographic separation was achieved on a Zorbax SB-C18 column (150×4.6 mm id, 5 μm; Agilent, Santa Clara, CA, USA) under isocratic conditions. The mobile phase was 5 mmol/L ammonium acetate/methanol/acetonitrile/formic acid (20:27:53:0.1, v/v), and the flow rate was 0.6 mL/min. The column temperature was 45 °C. The mass spectrometer was operated in the positive mode with an ion spray voltage of 4200 V and a source temperature of 500 °C. The nebulizer gas, heater gas, collision gas, and curtain gas were set at 50, 50, 4, and 10 psi, respectively. The declustering potential and entrance potential were maintained at 40 and 61 V, respectively. The mass transitions for multiple reaction monitoring were m/z 613.3 → m/z 511.3 and 537.3 for HMME, and m/z 616.3 → m/z 511.3 and 537.3 for d3-HMME (internal standard). The collision energy for all transitions was set to 25 eV.
Data analysis
GraphPad Prism (version 6.01, GraphPad Software Inc, San Diego, CA, USA) was used to calculate the uptake kinetic parameters and the half inhibitory concentration (IC50). Phoenix WinNonlin (version 6.1, Pharsight Corp, Cary, NC, USA) was used to calculate the pharmacokinetic parameters with a non-compartmental model. All data are expressed as the mean±standard deviation (SD).
The substrate uptake rate was normalized to the protein concentration of the cell lysate, and the transporter-mediated uptake was corrected by subtracting the accumulation in the mock-transfected HEK293 cells (or hepatic uptake at 4 °C) from that in the OATP-expressing HEK293 cells (or hepatic uptake at 37 °C).
Uptake kinetics
For the kinetic parameter calculations, the data were fitted to the Michaelis-Menten equation (1) or allosteric sigmoidal modeling (2) according to the Eadie-Hofstee plots.


where V (pmol·min−1·mg−1) is the velocity of substrate uptake, S (μmol/L) is the substrate concentration, Km (μmol/L) is the substrate concentration at half-maximal uptake rate (Vmax), and h is the Hill slope. CLint is Vmax/Km for both models.
Inhibition study
For the inhibition study, the IC50 values were obtained by plotting the log inhibitor concentration against the normalized response according to the following equation (3):

where Y is the normalized response expressed as the percentage of the net uptake rate in the absence of inhibitors, and X is converted to the log inhibitor concentration (μmol/L).
SPSS software (version 16.0 for Windows, SPSS Inc, Chicago, IL, USA) was used to determine significant differences with Student's two-tailed unpaired t tests. P-values <0.05 were regarded as statistically significant.
Results
Effects of rifampicin on the pharmacokinetics of HMME in rats
First, the effects of rifampicin, a specific Oatps inhibitor, on the pharmacokinetics of HMME isomers were examined (Figure 2, Table 1). After an intravenous injection of 5 mg/kg of HMME, HMME was quickly eliminated with a mean residence time (MRT) of approximately 0.39 h. HMME-1 and HMME-2 had different pharmacokinetic profiles; the plasma concentration of HMME-1 was 1.5 to 3.7 times higher than that of HMME-2 over the entire collection time. Co-administration of rifampicin increased the maximum concentrations (Cmax) of HMME-1 and HMME-2 by 13% and 47%, respectively, and significantly decreased the Cmax ratio of HMME-1 to HMME-2 from 1.5 to 1.1. By contrast, the systemic exposures, expressed as the area under the curve (AUC), of HMME-1 and HMME-2 increased significantly by 69% and 115%, respectively, resulting in a decrease in the AUC ratio of HMME-1 to HMME-2 from 1.98 to 1.56. Administration of rifampicin significantly decreased the clearances of HMME-1 and HMME-2 by 40% to 50%. These data suggest that Oatps-mediated hepatic uptake may be involved in the selective elimination of HMME isomers in rats.

Effects of rifampicin (25 mg/kg, intravenous) on the plasma concentration-time curves of HMME-1 and HMME-2 in rats after intravenous administration of 5 mg/kg HMME. The data are shown as the mean±SD of quadruplicates.
Uptake of HMME into OATP-transfected HEK293 cells
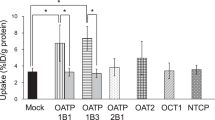
The uptake properties of HMME in the OATP-expressing HEK293 cells were also determined. We have previously validated the function of OATP transporters using typical substrates (E3S for OATP1B1 and OATP2B1 and BSP for OATP1B3) (data not shown). The uptake of both HMME isomers was significantly greater in the OATP1B1-, OATP1B3-, and OATP2B1-expressing HEK293 cells, compared with the mock-transfected HEK293 cells (Figure 3). Administration of 200 μmol/L rifampicin completely abolished OATP1B1- and OATP1B3-mediated uptake of HMME but did not affect OATP2B1-mediated uptake. Similar results were obtained for the inhibitors BSP and cyclosporine (data not shown). Telmisartan (50 μmol/L) almost completely inhibited HMME uptake by all three OATP transporters. Because the uptake-time profile of 2 μmol/L HMME revealed that both HMME isomers were rapidly transported into the transporter-expressing cells (Figure 4), 30 s was chosen as the uptake time for the subsequent concentration-dependent assay. Instead of the typical Michaelis–Menten curve, OATP1B1- and OATP2B1-mediated HMME-1 uptake displayed a sigmoidal autoactivation profile, and OATP1B3-mediated HMME-1 uptake and OATP1B1-mediated HMME-2 uptake exhibited biphasic kinetics (Figure 5A and 5B). The Km and Vmax values of HMME-1 and HMME-2 are summarized in Table 2. OATP1B1 and OATP1B3 exhibited greater affinities for HMME-2 (Km values of 0.63 and 0.58 μmol/L, respectively) than for HMME-1 (Km values of 5.61 and 1.51 μmol/L, respectively), whereas OATP2B1 exhibited similar affinities towards HMME-1 and HMME-2 (Km values of 1.48 and 1.92 μmol/L, respectively). When HEK293 cells were exposed to HMME over a concentration range of 0.05–10 μmol/L, HMME-2 uptake was approximately 2.3 to 6.6 times greater than that of HMME-1 in the OATP1B1- and OATP2B1-expressing HEK293 cells. However, OATP1B3-mediated HMME-1 uptake was 1.5 times greater than that of HMME-2, when the concentration of HMME was greater than 2 μmol/L (Figure 5C).

Cellular uptake of HMME-1 (A) and HMME-2 (B) into OATP- or mock-transfected HEK293 cells. The uptake of 2 μmol/L HMME was determined in the absence or presence of rifampicin (200 μmol/L) or telmisartan (50 μmol/L) for 10 min. Mean±SD. n=3. bP<0.05, cP<0.01 versus mock or control values.

Uptake-time profiles of HMME-1 (A) and HMME-2 (B) into OATP- and mock-transfected HEK293 cells. The substrate concentration of HMME was 2 μmol/L. Mean±SD. n=3. OATP-mediated HMME-1 and HMME-2 uptake was significantly (P<0.05) greater than that of mock-mediated uptake.

Concentration-dependent uptake of HMME-1 (A), HMME-2 (B), and HMME (C) into OATP-expressing HEK293 cells. The uptake time was 30 s. (A, B) The concentration-dependent uptake of each HMME isomer was represented by a nonlinear fitting curve and Eadie-Hofstee plots. (C) Accumulation of HMME-1 (circles) and HMME-2 (squares) in HEK293 cells were determined by exposing HMME to cells. Transporter-mediated HMME accumulation was corrected by subtracting the nonspecific accumulation in the mock-transfected HEK293 cells from that of the OATP-expressing HEK293 cells. Mean±SD. n=3. bP<0.05, cP<0.01 versus HMME-1 values.
Uptake of HMME into isolated rat hepatocytes
The uptake of HMME into isolated rat hepatocytes was time- and temperature-dependent. The uptake of both isomers at 37 °C was 3 to 7 times greater than that at 4 °C, indicating that active transporters are involved in HMME uptake into rat hepatocytes (Figure 6A and 6B). The uptake–time profiles of both HMME isomers exhibited good linearity for 10 min; however, the transporter-mediated uptake of HMME-2 was greater than that of HMME-1 for incubation time up to 2 min (Figure 6C). Because HMME was rapidly cleared from the circulation in rats, an incubation time of 30 s was used for further kinetic studies. The transporters involved in HMME uptake into rat hepatocytes were studied over 10 min using selective transporter inhibitors at 37 °C (Figure 6D). The uptake of HMME-1 and HMME-2 into rat hepatocytes was decreased by 82.0% and 81.8%, respectively, in the presence of 50 μmol/L rifampicin (Oatps inhibitor). Ketoprofen (Oat2 inhibitor) and MPP+ (Oct1 inhibitor) inhibited the uptake of HMME-1 by 28.4% and 28.6%, respectively, and that of HMME-2 by 43.8% and 47.1%, respectively.

Uptake of HMME-1 and HMME-2 into rat hepatocytes. The concentration of HMME was 0.5 μmol/L. (A, B) The uptake of HMME-1 (A) and HMME-2 (B) into rat hepatocytes were determined for various times at 37 °C (squares) and 4 °C (circles). (C) The net uptake of HMME-1 and HMME-2 were calculated by subtracting the uptake of HMME isomers at 4 °C from that of 37 °C. (D) Inhibitory effects of 50 μmol/L rifampicin (Oatps inhibitor), 10 μmol/L ketoprofen (Oat2 inhibitor), and 10 μmol/L MPP+ (Oct1 inhibitor) on the uptake of HMME-1 and HMME-2 at 37 °C were investigated. Mean±SD. n=3. bP<0.05, cP<0.01 versus control values.
The concentration-dependent uptake of HMME or a single isomer into rat hepatocytes was determined over a concentration range of 0.1–15 μmol/L (Figure 7, Table 2). The Eadie–Hofstee plots for HMME-1 and HMME-2 exhibited sigmoidal and biphasic kinetic profiles, respectively. The Km and Vmax values for HMME-1 were 0.63±0.04 μmol/L and 64.8±2.2 pmol·min−1·mg−1, respectively, calculated using the allosteric sigmoidal equation, and the same values for HMME-2 were 3.77±0.85 μmol/L and 269±33 pmol·min−1·mg−1, respectively, calculated using the Michaelis–Menten equation. In rat hepatocytes exposed to HMME, the accumulation of HMME-2 was approximately 1.5 times greater than that of HMME-1 over a concentration range of 1–15 μmol/L of HMME.

Concentration-dependent uptake of HMME-1 (A), HMME-2 (B), and HMME (C) into rat hepatocytes. The uptake time was 30 s. (A, B) The concentration dependence of the uptake of a single HMME isomer was represented by a nonlinear fitting curve and Eadie-Hofstee plots. (C) The accumulation of HMME-1 (circles) and HMME-2 (squares) in the hepatocytes were determined by exposing HMME to cells. Transporter-mediated HMME accumulation was corrected by subtracting the nonspecific accumulation in rat hepatocytes at 4 °C from that at 37 °C. Mean±SD. n=3. bP<0.05, cP<0.01 versus HMME-1 values.
Uptake of HMME into human cryopreserved hepatocytes
HMME was taken up by human hepatocytes (lot LTG) in a time- and temperature-dependent manner (Figure 8A). The uptake of both isomers at 37 °C was 2.7–3.9 times higher than that at 4 °C, indicating that active transporters are involved in HMME uptake into human hepatocytes. The identity of the transporters involved in HMME human hepatic uptake was investigated using selective transporter inhibitors (Figure 8B). Ketoprofen (OAT2 inhibitor) and MPP+ (OCT1 inhibitor) did not affect the uptake rates of HMME-1 and HMME-2, but the uptake of HMME-1 and HMME-2 was significantly decreased by approximately 43.8% and 46.4%, respectively, in the presence of 50 μmol/L rifampicin (OATPs inhibitor).

Uptake of HMME-1 and HMME-2 into adherent human hepatocytes (Lot LTG) in the absence or presence of transporter inhibitors. (A) The hepatic uptake of 2 μmol/L HMME were determined for 1 and 5 min at 37 °C and 4 °C. (B) Inhibitory effects of rifampicin (50 μmol/L, OATPs inhibitor), ketoprofen (10 μmol/L, OAT2 inhibitor), and MPP+ (10 μmol/L, OCT1 inhibitor) on the hepatic uptake of 2 μmol/L HMME was investigated at 37 °C for 10 min. Mean±SD. n=3. bP<0.05, cP<0.01 versus control values.
Uptake kinetic studies of individual HMME isomers into human hepatocytes (lot YJM) were conducted over a concentration range of 0.1–15 μmol/L. The Eadie-Hofstee plots for HMME-1 and HMME-2 contained one saturable component (Figure 9). The Km and Vmax values of the single isomers were 4.45±1.05 μmol/L and 50.3±4.6 pmol·min−1·mg−1, respectively, for HMME-1 and were 0.98±0.08 μmol/L and 62.8±1.6 pmol·min−1·mg−1, respectively, for HMME-2 (Table 2). The uptake of HMME-2 into hepatocytes (lot YJM) was approximately 3.9 times greater than that of HMME-1 over a concentration range of 0.2–15 μmol/L of HMME, and in addition, the uptake of HMME-2 was significantly greater than that of HMME-1 in hepatocytes from the lots LTG and HC2-25 (data not shown).

Concentration-dependent uptake of HMME-1 (A), HMME-2 (B), and HMME (C) into human hepatocytes. The uptake time was 1 min. (A, B) The concentration dependence of the uptake of a single HMME isomer was represented by a nonlinear fitting curve and Eadie–Hofstee plots. (C) The accumulation of HMME-1 (circles) and HMME-2 (squares) in the hepatocytes were determined by exposing HMME to hepatocytes. Transporter-mediated HMME accumulation was corrected by subtracting the nonspecific accumulation in human hepatocytes at 4 °C from that at 37 °C. Mean±SD. n=3. bP<0.05, cP<0.01 versus HMME-1 values.
Inhibitory effects of rifampicin and telmisartan on HMME uptake
The inhibitory effects of rifampicin and telmisartan on the uptake of HMME-1 and HMME-2 were investigated in human hepatocytes and OATP-expressing HEK293 cells. Telmisartan and rifampicin inhibited OATP1B1- and OATP1B3-mediated HMME uptake in a concentration-dependent manner with IC50 values between 1.18 to 32.8 μmol/L (Figure 10 and Table 3). Telmisartan, rather than rifampicin, strongly inhibited OATP2B1-mediated uptake of 1 μmol/L HMME with IC50 values of approximately 1.79 and 3.10 μmol/L for HMME-1 and HMME-2, respectively. In human hepatocytes, 50 μmol/L telmisartan inhibited approximately 75.7% of the HMME-1 uptake and 85.3% of the HMME-2 uptake. When the inhibitory effect of telmisartan on OATP-mediated HMME uptake in human hepatocytes was taken as 100%, rifampicin was observed to inhibit HMME-1 and HMME-2 uptake by 79.1% and 83.7%, respectively.

Inhibitory effects of rifampicin (left panel) and telmisartan (right panel) on the uptake of HMME mediated by OATP1B1 (A), OATP1B3 (B), and OATP2B1 (C). The HMME concentration was 1 μmol/L, and the uptake time was 30 s. Transporter-mediated HMME accumulation was corrected by subtracting the nonspecific accumulation in the mock-transfected HEK293 cells from that in the OATP-expressing HEK293 cells. Mean±SD. n=3.
Discussion
In this study, we demonstrated that Oatps-mediated rapid hepatic uptake is responsible for the rapid clearance of HMME from the circulation and for its selective pharmacokinetics in rats. Our study additionally showed that the hepatic uptake of HMME in humans is predominately mediated by OATP1B1 and that the greater uptake of HMME-2 by OATP1B1 may lead to a lower exposure of HMME-2 than HMME-1 in humans.
The preliminary studies showed that HMME was quickly eliminated unchanged through the bile/feces in rats and humans after an intravenous administration3,4, indicating that the liver is critical to HMME disposition. In the current study, the pharmacokinetic behaviors were observed to be different for the two HMME isomers. The systemic exposure of HMME-1 was double that of HMME-2 in rats (Table 1). To investigate the mechanisms involved in the hepatobiliary disposition of HMME, freshly isolated rat hepatocytes were used to evaluate the transport properties of HMME. Both HMME isomers were transported into rat hepatocytes in a temperature- and time-dependent manner (Figure 6). The uptake of HMME-2 was 1.5 times greater than that of HMME-1 over an HMME concentration range of 1–15 μmol/L, which covers the maximum free concentration (approximately 5.8–8.6 μmol/L) of HMME in rats and humans3,4. Consequently, a greater uptake of HMME-2 in rat hepatocytes should be observed immediately after a dose of HMME, and this may explain why the systemic exposure of HMME-2 is less than that of HMME-1. When HMME was co-administered with rifampicin (Oatps inhibitor) in rats, the Cmax and AUC values for the isomers increased by different amounts as the Cmax and the AUC ratios of HMME-1 to HMME-2 decreased. This indicates that Oatps play an important role in the selective hepatic elimination of HMME isomers. Oat2 and Oct1 may also be involved in the hepatic uptake of HMME in rats (Figure 6D).
Although Oatps and OATPs have overlapping substrate profiles, the differences in the expression and function of Oatps/OATPs may lead to different substrate disposition in rats and humans. Therefore, to investigate the hepatic disposition of HMME in humans, the transporters involved in HMME hepatic uptake were determined. Chemical inhibition studies in human hepatocytes and OATP-expressing HEK293 cells demonstrated that HMME is a substrate of OATP1B1, OATP1B3, and OATP2B1, but not of OAT2 or OCT1 (Figures 3 and 8B). Consequently, it was necessary to identify the major OATP isoforms involved in hepatic HMME uptake. Interestingly, OATPs showed different uptake characteristics for HMME-1 and HMME-2. HMME-2 uptake was approximately 2.3–6.9 times greater than that of HMME-1 in human hepatocytes and in OATP1B1- or OATP2B1-expressing HEK293 cells over an HMME concentration range of 0.2–15 μmol/L. However, OATP1B3-mediated HMME-1 uptake was approximately 1.5 times greater than that of HMME-2 over an HMME concentration range of 2–10 μmol/L. Considering the disposition of HMME-1 and HMME-2 in human hepatocytes and OATP-expressing HEK293 cells, OATP1B1 and OATP2B1 are probably the major transporters involved in HMME uptake. The Km values for OATP1B1-mediated HMME-1 and HMME-2 uptake were 5.61 and 0.63 μmol/L, respectively, which were comparable to the Km values in human hepatocytes. Unlike OATP1B1, OATP2B1 exhibited similar affinities towards HMME-1 and HMME-2 (Km values of 1.48 and 1.92 μmol/L, respectively), which suggests an indecisive role of OATP2B1 in the selective hepatic uptake of HMME. In addition, it was reported that the hepatic expression level of OATP2B1 is much lower than that of OATP1B1 and OATP1B317,18. Therefore, the transporter uptake and expression data suggest that OATP1B1 is most likely the predominant OATP isoform involved in HMME uptake into human hepatocytes.
OATP1B1 was identified as the major transporter for pitavastatin, according to the relative activity factor and relative expression factor9. Telmisartan was identified as a selective substrate of OATP1B3, and it was characterized using E3S as an OATP1B1-selective inhibitor19. Instead of the approaches estimating the relative contributions of transporters listed above, we used a non-specific OATP inhibitor and a non-OATP2B1 inhibitor to estimate the contribution of OATP2B1 to the HMME hepatic uptake. Rifampicin is a well-known inhibitor that inhibits OATPs in the order OATP1B3>OATP1B1>OATP2B114. In our experiments, rifampicin inhibited OATP1B1- and OATP1B3-mediated HMME uptake, but hardly inhibited OATP2B1. It was hypothesized that only when two drugs share the same binding site on OATP2B1 could DDI be observed, because there were multiple binding sites on OATP2B120. Therefore, the lack of inhibitory effects of rifampicin on the OATP2B1-mediated HMME uptake may be due to the different binding sites of rifampicin and HMME on OATP2B1. We also showed that telmisartan is a non-specific potent inhibitor for OATP1B1, OATP1B3, and OATP2B1 with stronger inhibitory effects than rifampicin. By comparing the different inhibitory effects of telmisartan and rifampicin on hepatic HMME uptake, we showed that OATP2B1 accounts for <20% of OATP-mediated HMME uptake into human hepatocytes (Table 3). We confirmed that OATP1B1, rather than OATP2B1, is the major transporter involved in hepatic HMME uptake, which is consistent with our hypothesis. Thus, the greater uptake of HMME-2 mediated by OATP1B1 could potentially lead to a lower systemic exposure of HMME-2, compared with HMME-1, in humans.
The rapid hepatic uptake of HMME facilitated the clearance of HMME from the circulation, which may reduce the risk of skin phototoxicity. Multiple genetic polymorphisms of OATP1B1 have been identified, and these polymorphisms may influence the pharmacokinetics of its substrates21. A common single nucleotide polymorphism, c.521T>C, which has a high frequency of approximately 12% in East Asians, decreases the hepatic expression of OATP1B1 and attenuates its transport activity for E3S and statins22. Compared with wild-type OATP1B1, c.521T>C was associated with increased systemic exposure to multiple drugs, including oral pravastatin23,24, fexofenadine25, and lopinavir26,27. In addition, the co-administration of OATP1B1 inhibitors may affect the pharmacokinetics of its substrates. For example, a single dose of rifampicin increased the AUC of glyburide by 225% and cyclosporine increased that of rosuvastatin by 710%28,29. Therefore, when the activity of OATP1B1 is impaired by genetic variation or co-administered inhibitors, subjects with lower OATP1B1 activity may have higher systemic exposure and a lower hepatic clearance of HMME, followed by an increased risk of skin phototoxicity. However, as a compensatory mechanism, OATP1B3 and OATP2B1 may transport HMME, promoting the clearance of HMME and alleviating the risk of skin phototoxicity. In this case, the hepatic disposition of HMME isomers could be altered by the different contributions of the transporters involved. It is noteworthy that OATP-mediated drug-drug interactions between telmisartan and HMME in clinic would be limited, as the free Cmax values of telmisartan (approximately 0.028 μmol/L) was much lower than its IC50 values for the OATP transporters19,30.
It has been accepted that breast cancer resistance protein (BCRP, ABCG2) is involved in the efflux of porphyrins from cells31,32,33,34. However, little is known about the involvement of uptake transporters in the in vivo disposition of porphyrins. Considering the poor passive diffusion of porphyrins, uptake transporters are most likely involved in the uptake of porphyrins from the circulation. Intestinal heme carrier protein 1 (SLC46A1), a member of the major facilitator superfamily, mediates the uptake of heme from plasma to the duodenum35; however, there is no evidence that this protein is involved in the hepatic uptake of porphyrins. A patient with variegate porphyria, which is characterized by elevated levels of circulating porphyrins, showed decreased elimination of indocyanine green and BSP, typical substrates for OATPs5,36. Porphyrins, such as hematoporphyrin and coporphyrin, with side chains that are negatively charged under physical conditions (pH 7.4) are potent inhibitors for OATP1B1-mediated estradiol 17β-D-glucuronide uptake37. The studies listed above suggest that porphyrins may be substrates of OATP1B1. HMME contains propionic acid side chains, which is similar to hematoporphyrin. In this study, we demonstrated that HMME is a substrate of OATP1B1, OATP1B3, and OATP2B1. Our work provides good evidence for the involvement of OATPs in the hepatic uptake of certain porphyrins with charged acidic side chains. However, OATP-mediated transport of porphyrins may be affected by several factors, including the dimerization equilibrium constant, presence of multicharged ions, and the bulky structure of the porphyrin37. Thereby, the question of whether OATPs are involved in porphyrin uptake in hepatocytes should be further investigated in OATP-expressing systems.
Our previous study revealed that most of the HMME was recovered in rat bile within 2 h of administering the dose, indicating that canalicular efflux transporters play a vital role in HMME excretion to bile. BCRP expressed on the apical membrane of hepatocytes contributed to the efflux of porphyrins to bile and maintained the intracellular porphyrin homeostasis33. The polymorphisms of ABCG2, which are deficient in porphyrins transport, may be related to porphyria32. However, P-glycoprotein (P-gp) has not been reported to be involved in porphyrins transport yet. In future studies, we will identify whether P-gp and BCRP are involved in HMME efflux into bile to fully describe the hepatobiliary elimination of HMME in vivo.
In summary, this study showed that OATP1B1 is the major OATP isoform involved in the rapid hepatic uptake of HMME and that greater uptake of HMME-2 mediated by OATP1B1 may reduce the systemic exposure of HMME-2, compared with that of HMME-1 in humans. Our findings suggest that OATP1B1-related polymorphisms or drug–drug interactions may affect the pharmacokinetics of HMME isomers. In addition, our results extend the understanding of the mechanisms of porphyrins uptake into hepatocytes.
Author contribution
Xiu-li LI and Da-fang ZHONG are responsible for the research design; Xiu-li LI, Zi-tao GUO, and Ye-dong WANG conducted the experiments; Da-fang ZHONG, Xiao-yan CHEN, and Jia LIU contributed new reagents or analytic tools; Xiu-li LI and Da-fang ZHONG performed the data analysis and wrote this manuscript.
References
Song K, Kong B, Li L, Yang Q, Wei Y, Qu X . Intraperitoneal photodynamic therapy for an ovarian cancer ascite model in Fischer 344 rat using hematoporphyrin monomethyl ether. Cancer Sci 2007; 98: 1959–64.
Cai H, Gu Y, Sun Q, Zeng J, Dong N, Zhao G . Effect of hematoporphyrin monomethyl ether-mediated photodynamic therapy on hypertrophic scar fibroblasts. Photodermatol Photoimmunol Photomed 2011; 27: 90–6.
Pu Y, Chen W, Yu Z . Research progress of Hemoporfin — part one: preclinical study. Photodiagnosis Photodyn Ther 2012; 9: 180–5.
Sun PH, Zhao X, Zhou Y, Liang Y, Zhang HL, Cui YM, et al. Tolerance and pharmacokinetics of single-dose intravenous hemoporfin in healthy volunteers. Acta Pharmacol Sin 2011; 32: 1549–54.
Cui Y, Konig J, Leier I, Buchholz U, Keppler D . Hepatic uptake of bilirubin and its conjugates by the human organic anion transporter SLC21A6. J Biol Chem 2001; 276: 9626–30.
Treiber A, Schneiter R, Hausler S, Stieger B . Bosentan is a substrate of human OATP1B1 and OATP1B3: Inhibition of hepatic uptake as the common mechanism of its interactions with cyclosporin A, rifampicin, and sildenafil. Drug Metab Dispos 2007; 35: 1400–7.
Treiber A, Schneiter R, Delahaye S, Clozel M . Inhibition of organic anion transporting polypeptide-mediated hepatic uptake is the major determinant in the pharmacokinetic interaction between bosentan and cyclosporin A in the rat. J Pharmacol Exp Ther 2004; 308: 1121–9.
Shitara Y, Itoh T, Sato H, Li AP, Sugiyama Y . Inhibition of transporter-mediated hepatic uptake as a mechanism for drug-drug interaction between cerivastatin and cyclosporin A. J Pharmacol Exp Ther 2003; 304: 610–6.
Hirano M, Maeda K, Shitara Y, Sugiyama Y . Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J Pharmacol Exp Ther 2004; 311: 139–46.
Noe J, Portmann R, Brun ME, Funk C . Substrate-dependent drug-drug interactions between gemfibrozil, fluvastatin and other organic anion-transporting peptide (OATP) substrates on OATP1B1, OATP2B1, and OATP1B3. Drug Metab Dispos 2007; 35: 1308–14.
Gao CY, Zhang HJ, Guo ZT, You TG, Chen XY, Zhong DF . Mechanistic studies on the absorption and disposition of scutellarin in humans: selective OATP2B1-mediated hepatic uptake is a likely key determinant for its unique pharmacokinetic characteristics. Drug Metab Dispos 2012; 40: 2009–20.
Yamazaki M, Suzuki H, Hanano M, Tokui T, Komai T, Sugiyama Y . Na+-independent multispecific anion transporter mediates active-transport of pravastatin into rat-liver. Am J Physiol 1993; 264: G36–44.
Khamdang S, Takeda M, Noshiro R, Narikawa S, Enomoto A, Anzai N, et al. Interactions of human organic anion transporters and human organic cation transporters with nonsteroidal anti-inflammatory drugs. J Pharmacol Exp Ther 2002; 303: 534–9.
Vavricka SR, Van Montfoort J, Ha HR, Meier PJ, Fattinger K . Interactions of rifamycin SV and rifampicin with organic anion uptake systems of human liver. Hepatology 2002; 36: 164–72.
Tirona RG, Leake BF, Wolkoff AW, Kim RB . Human organic anion transporting polypeptide-C (SLC21A6) is a major determinant of rifampin-mediated pregnane X receptor activation. J Pharmacol Exp Ther 2003; 304: 223–8.
Lee WK, Reichold M, Edemir B, Ciarimboli G, Warth R, Koepsell H, et al. Organic cation transporters OCT1, 2, and 3 mediate high-affinity transport of the mutagenic vital dye ethidium in the kidney proximal tubule. Am J Physiol Renal Physiol 2009; 296: F1504–13.
Hirano M, Maeda K, Shitara Y, Sugiyama Y . Drug-drug interaction between pitavastatin and various drugs via OATP1B1. Drug Metab Dispos 2006; 34: 1229–36.
Keitel V, Burdelski M, Warskulat U, Kuhlkamp T, Keppler D, Haussinger D, et al. Expression and localization of hepatobiliary transport proteins in progressive familial intrahepatic cholestasis. Hepatology 2005; 41: 1160–72.
Ishiguro N, Maeda K, Kishimoto W, Saito A, Harada A, Ebner T, et al. Predominant contribution of OATP1B3 to the hepatic uptake of telmisartan, an angiotensin II receptor antagonist, in humans. Drug Metab Dispos 2006; 34: 1109–15.
Shirasaka Y, Mori T, Shichiri M, Nakanishi T, Tamai I . Functional pleiotropy of organic anion transporting polypeptide OATP2B1 due to multiple binding sites. Drug Metab Pharmacokinet 2012; 27: 360–4.
Tirona RG, Leake BF, Merino G, Kim RB . Polymorphisms in OATP-C: identification of multiple allelic variants associated with altered transport activity among European- and African-Americans. J Biol Chem 2001; 276: 35669–75.
Kameyama Y, Yamashita K, Kobayashi K, Hosokawa M, Chiba K . Functional characterization of SLCO1B1 (OATP-C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15+C1007G, by using transient expression systems of HeLa and HEK293 cells. Pharmacogenet Genomics 2005; 15: 513–22.
Nishizato Y, Ieiri I, Suzuki H, Kimura M, Kawabata K, Hirota T, et al. Polymorphisms of OATP-C (SLC21A6) and OAT3 (SLC22A8) genes: consequences for pravastatin pharmacokinetics. Clin Pharmacol Ther 2003; 73: 554–65.
Niemi M, Schaeffeler E, Lang T, Fromm MF, Neuvonen M, Kyrklund C, et al. High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SLCO1B1). Pharmacogenetics 2004; 14: 429–40.
Niemi M, Kivisto KT, Hofmann U, Schwab M, Eichelbaum M, Fromm MF . Fexofenadine pharmacokinetics are associated with a polymorphism of the SLCO1B1 gene (encoding OATP1B1). Br J Clin Pharmacol 2005; 59: 602–4.
Hartkoorn RC, Kwan WS, Shallcross V, Chaikan A, Liptrott N, Egan D, et al. HIV protease inhibitors are substrates for OATP1A2, OATP1B1 and OATP1B3 and lopinavir plasma concentrations are influenced by SLCO1B1 polymorphisms. Pharmacogenet Genom 2010; 20: 112–20.
Kohlrausch FB, de Cassia Estrela R, Barroso PF, Suarez-Kurtz G . The impact of SLCO1B1 polymorphisms on the plasma concentration of lopinavir and ritonavir in HIV-infected men. Br J Clin Pharmacol 2010; 69: 95–8.
Simonson SG, Raza A, Martin PD, Mitchell PD, Jarcho JA, Brown CD, et al. Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin Pharmacol Ther 2004; 76: 167–77.
Zheng HX, Huang Y, Frassetto LA, Benet LZ . Elucidating rifampin's inducing and inhibiting effects on glyburide pharmacokinetics and blood glucose in healthy volunteers: unmasking the differential effects of enzyme induction and transporter inhibition for a drug and its primary metabolite. Clin Pharmacol Ther 2009; 85: 78–85.
Stangier J, Su CA, Roth W . Pharmacokinetics of orally and intravenously administered telmisartan in healthy young and elderly volunteers and in hypertensive patients. J Int Med Res 2000; 28: 149–67.
Robey RW, Steadman K, Polgar O, Bates SE . ABCG2-mediated transport of photosensitizers: potential impact on photodynamic therapy. Cancer Biol Ther 2005; 4: 187–94.
Tamura A, Watanabe M, Saito H, Nakagawa H, Kamachi T, Okura I, et al. Functional validation of the genetic polymorphisms of human ATP-binding cassette (ABC) transporter ABCG2: identification of alleles that are defective in porphyrin transport. Mol Pharmacol 2006; 70: 287–96.
Krishnamurthy P, Xie T, Schuetz JD . The role of transporters in cellular heme and porphyrin homeostasis. Pharmacol Ther 2007; 114: 345–58.
Bebes A, Nagy T, Bata-Csorgo Z, Kemeny L, Dobozy A, Szell M . Specific inhibition of the ABCG2 transporter could improve the efficacy of photodynamic therapy. J Photochem Photobiol B 2011; 105: 162–6.
Shayeghi M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N, et al. Identification of an intestinal heme transporter. Cell 2005; 122: 789–801.
Aizawa T, Hiramatsu K, Ohtsuka H, Niwa A, Yukimura Y, Yamada T, et al. Defective hepatic anion transport in variegate porphyria. Am J Gastroenterol 1987; 82: 1180–5.
Campbell SD, Lau WF, Xu JJ . Interaction of porphyrins with human organic anion transporting polypeptide 1B1. Chem Biol Interact 2009; 182: 45–51.
Acknowledgements
We greatly appreciate the help of Dr Xiao-qing WANG from Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences (Shanghai, China), for the chemical synthesis of d3-HMME and Dr Cen XIE from Shanghai Institute of Materia Medica, Chinese Academy of Sciences (Shanghai, China), for helpful discussions.
This work was supported by the National Natural Science Foundation of China (Grant 81173117).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, Xl., Guo, Zt., Wang, Yd. et al. Potential role of organic anion transporting polypeptide 1B1 (OATP1B1) in the selective hepatic uptake of hematoporphyrin monomethyl ether isomers. Acta Pharmacol Sin 36, 268–280 (2015). https://doi.org/10.1038/aps.2014.104
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2014.104
