
Abstract
Aim:
To investigate the effects of M3, a derivative of huperzine A, on the apoptosis induced by sodium nitroprusside (SNP) in PC12 cells.
Methods:
Cell viability was detected using MTT method. Apoptosis was examined with annexin V/prodium iodide (PI) stain. The levels of reactive oxygen species (ROS) were measured using fluorophotometric quantitation. The amount of malonaldehyde (MDA) was determined with MDA detection kits. The expression of caspase-3 and Hsp70 were analyzed using Western blotting.
Results:
Exposure of PC12 cells to SNP (200 μmol/L) for 24 h decreased the cell viability to 69.0% of that in the control group. Pretreatment with M3 (10 μmol/L) or huperzine A (10 μmol/L) significantly protected the cells against SNP-induced injury and apoptosis; the ratio of apoptotic bodies in PC12 cells was decreased from 27.3% to 15.0%. Pretreatment with M3 (10 μmol/L) significantly decreased ROS and MDA levels, and increased the expression of Hsp70 in the cells. Quercetin (10 μmol/L) blocked the protective effect of M3, while did not influence on that of huperzine A.
Conclusion:
M3 protects PC12 cells against SNP-induced apoptosis, possible due to ROS scavenging and Hsp70 induction.
Similar content being viewed by others

Introduction
Alzheimer's disease (AD) and Parkinson's disease (PD) are multifaceted, progressive neurodegenerative disorders that occur mainly in older age groups; however, the incidence of these diseases has increased in younger populations in recent years. Although these two disorders are characterized by different clinical manifestations and pathological phenomena, they are both categorized by a selective loss of neurons. Free radical-induced oxidative stress, mitochondrial dysfunction, and excitotoxic processes have been suggested to play a key role in the loss of neurons1, 2.
Hsp70, a member of the heat shock protein family, functions as an ATP-dependent molecular chaperone in protein folding, multi-protein complex assembly, transmembrane transport, and protein degradation3. This protein is also part of an inducible system that aids in cell survival by preventing and/or repairing stress-induced protein damage during and after detrimental environmental stresses, such as free-radical accumulation4. Pytlowany et al used PC12 cells to compare the protective effects of huperzine A (HupA) derivative M3 and HupA on sodium nitroprusside (SNP)-induced apoptosis5. PC12 cells are derived from a rat pheochromocytoma cell line and can acquire neuron-like properties, such as neurite extension, when exposed to nerve growth factor (NGF). These cells are often used to study PD because NGF-treated and untreated cells synthesize, store, secrete, and take up dopamine by processes that are similar to those of dopaminergic neurons6. PC12 cells are also used to study AD because they express β-amyloid (Aβ) precursor protein (APP), in keeping with the mRNA and protein levels found in cortical neurons7.
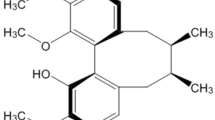
HupA, which is isolated from the herb Huperzia serrata, has been shown to be a highly effective, selective, and reversible acetylcholinesterase (AChE) inhibitor. The use of HupA as a therapeutic treatment for early or mild stages of AD may be due to additional mechanisms other than the reversible inhibition of AChE in the central nervous system. For example, HupA may target oxidative stress, Aβ-associated neurotoxicity, APP processing, and NGF8. The capacity of AChE to hydrolyze acetylcholine is not involved in the aforementioned mechanisms9. Therefore, we sought to identify derivatives of HupA that retain a similar level of AChE inhibition but yield an increase in cell protection. Due to the rigid molecular configuration of HupA, traditional chemical methods cannot be used to modify its structure. Instead, we chose to use microbial transformation to produce HupA derivatives. M3 is a product of the transformation of HupA by Streptomyces griseus CACC 200300 after a two-step procedure and various chromatographic techniques. The structure (Figure 1) was identified as huperzine A 8α,15α-epoxide using physical and chemical data collected through multiple analyses, such as HRMS, 1D NMR, 2D NMR, and IR. Although the IC50 value of M3 on AChE inhibition was established to be higher than that of HupA10, the remarkable effect of protecting PC12 cells from oxidative stress was observed in our present screening.

The structures of HupA and M3 by Streptomyces griseus CACC 200300.
Materials and methods
Materials and reagents
M3 was prepared as previously described10. HupA and SNP were bought from the National Institute of the Control of Pharmaceutical and Biological Products. Quercetin and Hoechst 33342 were purchased from Sigma. These compounds were dissolved in DMSO and diluted by PC12 medium. An annexin V/propidium iodide (PI) staining kit was bought from Beijing Biosea Biotechnology Company. Dichlorofluorescein diacetate (DCFH-DA) and MDA kits were bought from Beyotime Institute of Biotechnology. The anti-Hsp70 antibody and anti-mouse secondary antibody were bought from Santa Cruz Biotechnology. The anti-caspase-3 (17 kDa) antibody was bought from Cell Signaling Technology. The anti-β-actin antibody was bought from Sigma.
Cell culture and treatment
PC12 cells, purchased from the American Tissue Type Collection, were grown in 75-cm2 tissue culture flasks in DMEM supplemented with 5% heat-inactivated fetal bovine serum (FBS), 10% equine serum (ES), penicillin (100 IU/mL), streptomycin (100 mg/L), and L-glutamine (2 mmol/L). Cultures were maintained at 37 °C in 95% air-5% CO2 in a humidified incubator and passaged every 3 to 4 d. PC12 cells in logarithmic phase were seeded into 96-well or 6-well plates coated with 0.1 mg/mL poly-L-lysine (Sigma) and allowed to grow for at least 24 h. Different concentrations of M3 and HupA (0.1, 1, and 10 μmol/L) were added to the culture media for 1 h followed by addition of SNP (200 μmol/L) for 24 h. In some experiments, PC12 cells were pretreated with 10 μmol/L quercetin and 10 μmol/L M3 for 1 h and were exposed to 200 μmol/L SNP for the next 24 h.
MTT assay
PC12 cells were cultured at a density of 6000 cells per well in 96-well plates. MTT (5 mg/mL) was added to each well after the aforementioned treatments and the plates were incubated for 4 h. After centrifugation, the supernatant was removed from each well, and DMSO (100 μL) was added. After 30 min, the absorbance was recorded with a microplate reader (Bio-Rad model 550) at a wavelength of 570 nm. Each experiment was performed in triplicate.
Detection of apoptosis
PC12 cells were cultured at a density of 30 000 cells per well in 6-well plates. Cells were harvested by being digested in 0.25% trypsin after treatment, stained with annexin V for 20 min, and PI for another 10 min at 37 °C before detection using flow cytometry (EPICS XL, Beckman Coulter, USA). To stain the DNA, cells were incubated with Hoechst 33342 (10 μg/mL) diluted in phosphate-buffered saline (PBS) for 10 min at room temperature. Cells were then observed by the IN Cell Analyzer 1000 (GE Healthcare, USA).
Measurement of reactive oxygen species (ROS)
Intracellular accumulation of ROS was measured using fluorophotometric quantitation. The cells were cultured with M3 or HupA (10 μmol/L) for 1 h before exposure to SNP (200 μmol/L) for 24 h. The harvested cells were subsequently stained with DCFH-DA for 30 min at 37 °C. The cell suspension was dispensed into 96-well black plates. DCFH-DA reacts with ROS and is converted to dichlorofluorescein (DCF). The fluorescence from the DCF was analyzed using a Spectra microplate fluorescence reader (Max Gemini XS, MD, USA) with the excitation wavelength set at 488 nm and the emission wavelength set at 525 nm.
Estimation of lipid peroxidation
MDA reacts with thiobarbituric acid (TBA) to produce a fluorescent product. Levels of MDA were measured in PC12 cell lysates with a microplate reader at a wavelength of 535 nm. PC12 cells were treated with M3 1 h prior to exposure to SNP and left to grow to more than 90% confluence in 75-cm2 plates. Cells were harvested and washed with PBS after 24 h. The method described in the MDA detection kit was employed from Nanjing Jiancheng Bioengineering Institute, Nanjing, China.
Protein assay
After a 1-h treatment with M3 and 24 h exposure to SNP, cells were rapidly washed with PBS and lysed on ice in a lysis buffer (pH=8.0) containing 50 mmol/L Tris-HCl, 150 mmol/L NaCl, 1% NP-40, 1 mmol/L PMSF, 1 mmol/L EDTA, 50 mmol/L NaF, 1 mmol/L DTT, 1 mg/L aprotinin and 1% Triton X-100. Protein concentrations were measured with a BCA kit (Vigorous). The lysates were solubilized in an SDS sample buffer, separated with 15% SDS-PAGE, and transferred to PVDF membrane (Millipore). The membrane was blocked with 3% BSA and subsequently incubated with anti-Hsp70, anti-caspase-3 (17 kDa), or anti-β-actin antibodies followed by a horseradish peroxidase (HRP)-conjugated secondary antibody and detected with an ECL detection system (Molecular Device, Lmax). The density of each band was quantitated using image-analysis software (Science Lab 2005 Image Gauge; Fuji Film Co Ltd, Tokyo, Japan).
Statistical analysis
Data are expressed as the mean±standard deviation. An analysis of variance (ANOVA) followed by a Newman-Keuls post-hoc test was performed to assess the differences between groups. Values of P<0.05 were regarded as statistically significant.
Results
The neurotoxicity of SNP on PC12 cells
Before studying the protective effects of M3, we analyzed the toxicity of SNP on PC12 cells. PC12 cells treated with different concentrations of SNP for 24 h displayed a decrease in the production of formazan dye in a dose-dependent manner. Exposure to 200 μmol/L of SNP induced moderate injury to the cells when compared with the control group. This concentration of SNP was used to compare the neuroprotective effects of M3 and HupA (Figure 2).

Effect of SNP on the cell viability in PC12 cells. Cells were exposed to SNP for 24 h. The cell viability was evaluated by measuring the quantity of formazan generated by activated mitochondria. The values were expressed as percentage of control, which is set to 100%. The percentage of cell viability was presented as mean±SD for six replicates. bP<0.05 vs control group.
Pre-incubation with M3 and HupA protected PC12 cells from SNP-induced injury
Exposure to 200 μmol/L of SNP for 24 h decreased the viability of PC12 cells to 69.0%±4.8% compared with the control group. Pre-incubation with M3 or HupA for 1 h attenuated the injury induced by SNP in a dose-dependent manner. This effect was statistically significant at a concentration of 10 μmol/L (P<0.05). The cell viability after pre-incubation with M3 was 84.1%±2.7% of the control, while the cell viability after pre-incubation with HupA was 77.1%±3.5% of the control (P<0.05, Figure 3). There was no significant difference in cell viability between the M3 and HupA pre-treated groups (P>0.05).

Effect of HupA and M3 on the cell viability in PC12 cells induced by SNP. PC12 cells were pre-treated with HupA and M3 at different concertrations for 1 h before 200 μmol/L SNP was added. The percentage of cell viability was presented as mean±SD for six replicates. bP<0.05 vs control group. eP<0.05 vs SNP group.
M3 and HupA attenuated the SNP-induced apoptosis of PC12 cells
Analysis of an annexin V/PI stain using flow cytometry and Hoechst 33342 stain with a fluorescence microscope revealed that pretreatment with HupA or M3 significantly decreased the apoptosis of PC12 cells induced by SNP. M3 pretreatment alone showed no effect on apoptotic bodies or the nuclear morphology of PC12 cells. Pre-treatment with HupA and M3 (10 μmol/L) attenuated nuclear fragmentation and chromatin condensation, and inhibited apoptosis at the early and intermediate stages of SNP exposure. There was no statistically significant difference in the percentage of apoptotic bodies between the M3 and HupA groups (Figure 4).

M3 protected PC12 cells from apoptosis induced by SNP. PC12 cells were pre-treated with different doses of M3 for 1 h before 200 μmol/L SNP was added. (A) Cells were stained by annexin V and PI, and detected by flow cytometry. (a) control group; (b) 10 μmol/L M3 group; (c) 200 μmol/L SNP group; (d) 200 μmol/L SNP+10 μmol/L HupA group; (e) 200 μmol/L SNP+0.1 μmol/L M3 group; (f) 200 μmol/L SNP+1 μmol/L M3 group; (g) 200 μmol/L SNP+10 μmol/L M3 group. The percentage of apoptotic body was presented as mean±SD for three individual experiments. bP<0.05 vs control group. eP<0.05 vs SNP group. (B) Cells were stained by Hoechst 33342. The arrow indicates nuclear fragmentation or chromatin condensation.
M3 inhibited the production of SNP-induced ROS in PC12 cells
DCFH-DA is hydrolyzed to DCFH by intracellular esterases, which is subsequently oxidized by ROS to DCF, which is a fluorescent compound. The fluorescence intensity is directly proportional to the amount of ROS generated from SNP exposure. Using an excitation wavelength of 488 nm and an emission wavelength of 525 nm, the mean fluorescence intensity increased to 183.5%±18.3% in the SNP-treated group compared with the control group. Pre-incubation with 10 μmol/L of M3 or 10 μmol/L of HupA resulted in a decrease in the mean fluorescence intensity to 142.3%±20.2% or 132.0%±6.5%, respectively, compared with the SNP-treated group (Figure 5). There was no statistically significant difference in the levels of ROS between the M3 and HupA groups (P>0.05).

Effect of 10 μmol/L M3 on accumulation of ROS and MDA in PC12 cells induced by SNP. PC12 cells were pre-treated with 10 μmol/L M3 for 1 h before 200 μmol/L SNP was added. Cells were stained by DCFH-DA and detected by fluorometric analysis, or followed by the MDA detection kit. The percentage of ROS and MDA were presented as mean±standard deviation for at least three individual experiments. cP<0.01 vs control group. eP<0.05 vs SNP group.
Treatment with M3 decreased SNP-induced MDA accumulation in PC12 cells
MDA, formed by degradation of polyunsaturated lipids by ROS, is used as a marker to measure the level of oxidative stress in an organism. In our previous study, SNP increased MDA levels to 382.4%±57.2% compared with the control group. However, pre-incubation with 10 μmol/L of M3 or 10 μmol/L of HupA for 1 h caused a decrease in the levels of MDA to 288.7%±21.6% in the HupA pretreated group and 246.9%±44.2% in the M3 pretreated group (Figure 5). There was no statistically significant difference between the level of MDA in the M3 and HupA groups (P>0.05).
Treatment with M3 increased the expression of Hsp70 in PC12 cells
It was reported that an increase in the expression of Hsp70 confers an improvement in the survival ability of cells against SNP toxicity. The effect of M3 on the expression of Hsp70 in PC12 cells was analyzed in this study. The expression of Hsp70 increased in a time-dependent manner after M3 exposure, peaking after 0.5 h. No change was observed in Hsp70 expression after pre-incubation with HupA. After 1 h of exposure to SNP, the expression of Hsp70 increased. Pretreatment with M3 increased the Hsp70 level compared with cells exposed to SNP (Figure 6).

Effect of M3 on the expression of Hsp70 at different time. (A) Analysis of Hsp70 protein expression in PC12 cells by Western blotting at different time after exposure to M3 or HupA and the quantification of protein levels compare to the 0 h treating group by density scanning. bP<0.05 vs 0 h treating group. (B) Analysis of Hsp70 protein expression in PC12 cells by Western blotting after quercetin alone or with M3 co-application at different time and the quantification of protein levels compare to the 0 h treating group by density scanning. (C) Analysis of Hsp70 protein expression in PC12 cells with pretreatment of M3 at different time after exposure to SNP and the quantification of protein levels compare to the control group by density scanning. The percentage of density was presented as mean±standard deviation for at least three individual experiments. cP<0.01 vs control group. eP<0.05 vs SNP group.
Quercetin blocked the protective effect of M3 treatment on SNP-induced apoptosis in PC12 cells
Quercetin, a HSP biosynthesis inhibitor, was used to confirm if Hsp70 was involved in the protective effect of M3. As shown in Figure 7A, quercetin attenuated the protection conferred from M3 pretreatment on SNP-induced apoptosis while showing almost no effect on HupA-treated cells. As displayed in Figure 7B, treatment with HupA failed to significantly change the expression of Hsp70 compared with the expression of the SNP-exposed group. In M3 pretreated cells, the expression of Hsp70 increased to 212.5%±26.7% compared with control group. Caspase-3 expression remarkably increased in PC12 cells after exposure to SNP for 24 h, and pre-incubation with HupA and M3 for 1 h reversed this effect. Quercetin had no effect on the activation of caspase-3 in the control or HupA pretreated group, but it significantly increased the expression of caspase-3 in the M3 pretreated group (P<0.05).

Quercetin inhibited the protective effect of M3 against SNP-induced apoptosis in PC12 cells. PC12 cells were pre-treated with 10 μmol/L HupA, M3, or 10 μmol/L quercetin for 1 h then exposed to 200 μmol/L SNP for the next 24 h. (A) Effect of quercetin on the cell viability in PC12 cells by MTT method. The percentage of cell viability was presented as mean±standard deviation for six replicates. (B) Effect of quercetin on the ratio of Caspase-3 and Hsp70 in PC12 cells. The percentage of density was presented as mean±standard deviation for three individual experiments. cP<0.01 vs control group. eP<0.05 vs SNP group. hP<0.05 vs M3 pretreated group.
Discussion
Oxidative stress is recognized as an important mechanism in several neurodegenerative diseases. It has been proven that the activity of inducible nitric oxide synthases (iNOS) was increased in the brains of AD patients11. Free-radical production and nitration of endogenous biomolecules are the two main reasons that accumulation of nitric oxide (NO) induces cell loss12. In this study, SNP was used to induce the apoptosis of PC12 cells in order to evaluate potential neuroprotective compounds. Exposure to SNP in vitro could partially mimic the oxidative stress observed in the brains of neurodegenerative patients5. We found that, similar to the effect of HupA, pretreatment with M3 attenuated the apoptosis induced by SNP in PC12 cells. Although M3 and HupA showed a similar level of ROS scavenging, it seems that this was not the only mechanism involved in the protective effects of M3. Indeed, treatment with M3 induced the expression of Hsp70 in a time-dependent manner, unlike HupA. Hsp70, a member of the heat shock protein family, functions as a chaperone in protein folding, which can reduce abnormal protein aggregation13, 14. Overproduction of Hsp70 was shown to strongly protect brain cells from ischemia/reperfusion injuries both in vitro and in vivo15. Some of the anti-apoptotic activity of Hsp70 could be attributed to its ability to suppress the activity of JUN-kinase or Bid proteins16, 17. Thus, the decreased activation of caspase-3 in the M3 treated group might be due to the inhibition of the JNK/Bim pathway by the induction of Hsp70.
NO exposure has been established to induce Hsp70 expression in order to protect cells from tumor necrosis factor-induced apoptosis18. In our study, expression of Hsp70 could not save the PC12 cells from SNP-induced apoptosis. However, the M3 pretreatment remarkably increased the Hsp70 response without causing detrimental effects.
To confirm whether the induction of Hsp70 by M3 was responsible for its protective effects, quercetin, which is an HSP biosynthesis inhibitor, was used. Our results showed that pretreatment with quercetin attenuated the protective effect of M3 against SNP-induced apoptosis. This finding suggests that the protective effect of M3 was mediated, at least partly, by increasing the expression of Hsp70. However, quercetin failed to attenuate the protection of HupA on SNP-induced apoptosis, suggesting that the protective mechanism of HupA might have little interaction with Hsp70. Therefore, it appears that the protective mechanisms for these two compounds are different.
Taken together, M3 was similar to HupA with respect to free-radical scavenging. However, M3 treatment was able to elevate the expression of Hsp70 and decrease the expression of active caspase-3. M3 may activate the stress response without causing detrimental effects, thereby allowing the cells to survive under lethal conditions. Pretreatment with HupA also displayed ROS scavenging in PC12 cells exposed to SNP, but the presence of other protective mechanisms is not clear. Our research suggests that the distinctive chemical structure of HupA and its derivatives may be related to its anti-apoptotic properties. Further studies will concentrate on optimizing and analyzing these chemical structures in order to facilitate the design of novel anti-neurodegenerative drugs.
Author contribution
Na NING, Jin-feng HU, Yu-he YUAN, and Nai-hong CHEN developed the research plan; Na NING performed the experiments; Xin-yuan ZHANG and Jun-gui DAI contributed reagents or analytical tools; Na NING and Nai-hong CHEN analyzed the data; and Na NING prepared the manuscript.
References
Abdulwahid Al, Ahmad KH . Environmental toxins and Parkinson's disease: putative roles of impaired electron transport chain and oxidative stress. Toxicol Ind Health 2010; 26: 121–8.
Bonda DJ, Wang X, Perry G, Smith MA, Zhu X . Mitochondrial dynamics in Alzheimer's disease: opportunities for future treatment strategies. Drugs Aging 2010; 27: 181–92.
Thirumalai D, Lorimer GH . Chaperonin mediated protein folding. Annu Rev Biophys Biomol Struct 2001; 30: 245–69.
Hartl FU . Molecular chaperones in cellular protein folding. Nature 1996; 381: 571–9.
Pytlowany M, Strosznajder JB, Jeśko H, Cakała M, Strosznajder RP . Molecular mechanism of PC12 cell death evoked by sodium nitroprusside, a nitric oxide donor. Acta Biochim Pol 2008; 55: 339–47.
Yoko Hirata . Manganese-induced apoptosis in PC12 cells. Neurotoxicol Teratol 2002; 24: 639–53.
Lahiri DK, Ge YW . Role of the APP promoter in Alzheimer's disease: cell type-specific expression of the β-amyloid precursor protein. Ann N Y Acad Sci 2004; 1030: 310–6.
Zhang HY, Tang XC . Neuroprotective effects of huperzine A: new therapeutic targets for neurodegenerative disease. Trends Pharmacol Sci 2006; 27: 619–25.
Zhang HY, Brimijoin S, Tang XC . Apoptosis induced by beta-amyloid25–35 in acetylcholinesterase-overexpressing neuroblastoma cells. Acta Pharmacol Sin 2003; 24: 853–8.
Zhang XY, Zou JH, Dai JG . Microbial transformation of (–)-huperzine A. Tetrahedron Lett 2010; 51: 3840–2.
Malinski T . Nitric oxide and nitroxidative stress in Alzheimer's disease. J Alzheimers Dis 2007; 11: 207–18.
Imam SZ, el-Yazal J, Newport GD, Itzhak Y, Cadet JL . Slikker W Jr, et al Methamphetamine-induced dopaminergic neurotoxicity: role of peroxynitrite and neuroprotective role of antioxidants and peroxynitrite decomposition catalysts. Ann N Y Acad Sci 2001; 939: 366–80.
Hishiya A, Takayama S . Molecular chaperones as regulators of cell death. Oncogene 2008; 27: 6489–506.
Tutar L, Tutar Y . Heat shock proteins, an overview. Curr Pharm Biotechnol 2010; 11: 216–22.
Kabakov AE, Gabai VL . Heat-shock proteins maintain the viability of ATP-deprived cells: what is the mechanism? Trends Cell Biol 1994; 4: 193–6.
Gabai VL, Meriin AB, Yaglom JA, Volloch VZ, Sherman MY . Role of Hsp70 in regulation of stress-kinase JNK: implications in apoptosis and aging. FEBS Lett 1998; 438: 1–4.
Kumar Y, Tatu U . Stress protein flux during recovery from simulated ischemia: induced heat shock protein 70 confers cytoprotection by suppressing JNK activation and inhibiting apoptotic cell death. Proteomics 2003; 3: 513–26.
Kim YM, de Vera ME, Watkins SS, Billiar TR . Nitric oxide protects cultured rat hepatocytes from tumor necrosis factor induced apoptosis by inducing heat shock protein 70 expression. J Biol Chem 1997; 272: 1402–11.
Acknowledgements
This research was supported by the National Natural Sciences Foundation of China (No 81072541 and 30973887), and the Joint Funds of NSFC-Guangdong of China (No U0832008).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ning, N., Hu, Jf., Yuan, Yh. et al. Huperzine A derivative M3 protects PC12 cells against sodium nitroprusside-induced apoptosis. Acta Pharmacol Sin 33, 34–40 (2012). https://doi.org/10.1038/aps.2011.147
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2011.147
