
Abstract
Aim:
To investigate the effects of the natural product Withagulatin A on hepatic stellate cell (HSC) viability and type I procollagen production. The potential mechanism underlying the pharmacological actions was also explored.
Methods:
The effect of Withagulatin A on cell viability was evaluated in HSC and LX-2 cells using a sulforhodamine B (SRB) assay. Cell cycle distribution was analyzed using flow cytometry. Type I procollagen gene expression was determined using real-time PCR. Regulation of signaling molecules by Withagulatin A was detected using Western blotting.
Results:
Primary rat HSCs and the human hepatic stellate cell line LX-2 treated with Withagulatin A (0.625-20 μmol/L) underwent a dose-dependent decrease in cell viability, which was associated with S phase arrest and the induction of cell apoptosis. In addition, the natural product decreased phosphorylation of the Akt/mTOR/p70S6K pathway that controls cell proliferation and survival. Furthermore, Withagulatin A (1, 2 μmol/L) inhibited transforming growth factor-β (TGF-β) stimulated type I procollagen gene expression, which was attributable to the suppression of TGF-β stimulated Smad2 and Smad3 phosphorylation.
Conclusion:
Our results demonstrated that Withagulatin A potently inhibited HSC viability and type I procollagen production, thereby implying that this natural product has potential use in the development of anti-fibrogenic reagents for the treatment of hepatic fibrosis.
Similar content being viewed by others


Introduction
Hepatic fibrosis is considered a wound healing response to chronic liver injury1 and is caused by a variety of impairments such as HCV infection, alcohol abuse and non-alcoholic steatohepatitis (NASH)2. The hepatic stellate cell (HSC) has been established as the primary cell type responsible for liver fibrosis3. Moreover, activated HSCs undergo increased proliferation and overproduction of extracellular matrix (ECM) proteins4, thereby contributing to the progression of fibrosis. HSC activation is characterized by morphological changes marked by the appearance of the cytoskeletal protein α-smooth muscle actin (α-SMA)5 and is triggered by cytokines including mitogenic platelet-derived growth factor (PDGF) and transforming growth factor β1 (TGF-β1).
The phosphatidylinositol 3-kinase (PI3K)/Akt pathway controls a variety of cellular responses involving cell survival and proliferation6 and is strongly activated in HSCs by the potent mitogen PDGF7. Akt indirectly activates mammalian target of rapamycin (mTOR) through inhibition of TSC2 and directly phosphorylates mTOR at S24488. mTOR is a master regulator of protein synthesis and activates p70 ribosomal protein S6 kinase (p70S6K), which promotes protein translation9. Accordingly, inhibition of the mTOR/p70S6K pathway leads to reduced HSC proliferation10, which might serve as a potential anti-fibrotic strategy.
Type I collagen is a heterotrimer composed of two α1 chains and one α2 chain and is the prototypic constituent of the fibril-forming matrix in fibrotic liver. Activated HSCs are the principal cell type that produce type I collagen in response to TGF-β, which is the most potent stimulating factor for type I collagen gene transcription11. The Smad family proteins are essential regulators of TGF-β stimulated collagen gene transcription. Once activated by TGF-β, the receptor kinase TGF-β R1 phosphorylates Smad2 (Ser465/467) and Smad3 (Ser423/425) at their carboxy-termini. Phosphorylated Smad2 and Smad3 subsequently form a heteromeric complex with co-Smad and then translocate to the nucleus for DNA binding and transcriptional regulation12. To date, numerous studies have suggested the TGF-β/Smad pathway as promising anti-fibrotic targets13.
Physalis angulata L is an annual herb belonging to the Solanaceae family and is widely used in traditional Chinese medicine for the treatment of various illnesses including malaria, asthma, rheumatism and dermatitis. In addition, P angulata extract is clinically used for the treatment of human hepatic disorders including hepatitis14, 15. Withanolides are a group of structurally diverse steroids isolated from Physalis angulata and have been reported to exert varied pharmacological activities such as anticancer, immunosuppresive and antioxidant functions16. Withagulatin A is a withanolide from Physalis angulata and was demonstrated to inhibit proliferation of several tumor cell lines17. However, little is known concerning this compound's detailed pharmacological mechanism.
In the present study, we examined the pharmacological activity and underlying mechanisms for Withagulatin A (Figure 1A) mediated inhibition of liver fibrosis. We investigated whether Withagulatin A could inhibit HSC proliferation and tested its ability to induce cell apoptosis and cell cycle arrest, as well as its impact on the Akt/mTOR/p70S6K pathway. In addition, we examined whether Withagulation A could suppress TGF-β induced type I collagen expression and Smad2/3 phosphorylation. It is anticipated that our work will contribute to a better understanding of the pharmacological actions of Physalis angulata, and the natural product Withagulatin A will hopefully provide valuable structure information for the discovery of novel anti-liver fibrosis reagents.

Withagulatin A suppresses HSC activation and proliferation. (A) Chemical structure of Withagulatin A. (B) Withagulatin A suppresses α-SMA expression. Primary rat HSCs and human hepatic stellate LX-2cells were incubated with Withagulatin A (0, 1, and 2 μmol/L) for 24 h and the expression of α-SMA was determined by Western blotting. GAPDH was used as a loading control. The results shown are from three representative independent experiments. (C) Bands were quantified and data are expressed as fold of control. (D) Withagulatin A inhibited HSC proliferation. Primary rat HSCs, LX-2 cells, and LO2 hepatocytes were treated with a series of concentrations (0–20 μmol/L) of Withagulatin A for 48 h and cell viability was determined using a SRB assay. These experiments were carried out in triplicate. bP<0.05, cP<0.01 vs control cells (0 μmol/L).
Materials and methods
Reagents and antibodies
Withagulatin A was isolated from Physalis angulata L as previously described18 and was dissolved in dimethyl sulfoxide (DMSO) as a 2 mmol/L stock solution and stored at -20 °C. The DMSO concentration in all experiments was 0.1%. Sulforhodamine B (SRB) and all other chemicals were purchased from Sigma-Aldrich and were of analytical grade. Antibodies for phospho-Smad2 (Ser465/467), phospho-Smad3 (Ser423/425), Smad2, Smad3, phospho-CDK2, CDK2, Cyclin D1, and Cyclin B1 were purchased from Cell Signaling Technology. Antibodies for Bcl-2, Bax, and caspase3 were from Santa Cruz. The antibody for cleaved-caspase3 was purchased from Chemicon. The antibody for GAPDH was purchased from KangChen (Shanghai, China). The HRP-conjugated goat anti-rabbit and goat anti-mouse secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, USA).
Cell lines and cell cultures
Primary HSCs were isolated from normal livers of male Sprague-Dawley rats by a 2-step perfusion using pronase E and collagenase D (Sango, China), followed by nycodenz (Sigma-Aldrich) 2-layer discontinuous density gradient centrifugation as previously described19. All procedures in this experiment were performed according to the institutional ethical guidelines on animal care. The purity of rat HSC was assessed by autofluorescence at d 1. HSCs were cultured on uncoated plastic tissue culture dishes in DMEM supplemented with 20% heat inactivated fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin (Invitrogen), which were maintained in an incubator with a humidified atmosphere of 95% air and 5% CO2 at 37 °C. HSCs were activated by continuous culturing on uncoated plastic dishes for 7–10 d. Only fully transdifferentiated rat HSCs between d 10 and d 14 that exhibited the appropriate morphological changes were used for experiments to mimic the in vivo fibrotic state.
The human hepatic stellate cell line LX-2 was kindly provided by Dr Friedman (Mount Sinai School of Medicine, New York) and cultured according to previously report20. LO2 and LX-2 cells were cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin. CHO/EGFP-Smad2 cells were purchased from Thermo Scientific (Denmark), and cultured in F-12 medium supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin. Cells were maintained in an incubator with a humidified atmosphere of 95% air and 5% CO2 at 37 °C.
Cell viability assay
The effect of Withagulatin A on cell viability was determined using a SRB assay as previously reported21. Briefly, 1×104 cells/well were plated in 96-well plates for 24 h and then treated with varying concentrations of Withagulatin A for 24, 48, and 72 h. Cells were fixed, washed and stained with the SRB dye. Unbound dye was removed and the optical density was measured at 564 nm using a microplate spectrophotometer (Bio-Rad Laboratories). Cell viability was assessed as the percentage of viable cells compared to DMSO treated control cells, which were arbitrarily assigned as having 100% viability.
Apoptotic morphology
Apoptotic morphology was studied by staining the cells with Hoechst 33342 stain. Cells seeded on coverslips in a 6-well plate were treated with Withagulatin A (0, 1, and 2 μmol/L) for 24 h. The cover glasses were then washed with phosphate-buffered saline (PBS) and stained with 2 μmol/L of Hoechst 33342 for 15 min. Thereafter, the cells were washed with PBS and observed under a fluorescence microscope (Olympus). Nuclei condensation and fragmentation were considered cellular markers for an apoptotic morphology. Five fields were taken for each coverslip and the experiment was carried out in triplicate.
Flow cytometry assay
The effect of Withagulatin A on cell cycle progression was assessed by flow cytometry. Briefly, LX-2 cells at 70% confluence were treated with varying concentrations of Withagulatin A in complete medium for 24 h, trypsinized, washed, and fixed overnight with 70% ethanol at 4 °C. Cells were washed with PBS and incubated with 0.5 mg/mL RNaseA at 37 °C for 30 min and then incubated with 25 μg/mL propidium iodide on ice for 1 h in the dark. Cell cycle distribution was analyzed using a FACSCalibur instrument (BD Biosciences, San Jose, CA).
Western blot
Cells were lysed with lysis buffer containing 25 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 1 mmol/L Na3VO4, 1% Triton X-100 and a protease inhibitor cocktail (Sigma-Aldrich). Protein concentrations were determined using a BCA protein assay kit (Pierce, Rockford, IL). Equal amounts of lysates (30−40 μg protein) were resolved with 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Following electrophoresis, protein blots were transferred to a nitrocellulose membrane and probed with the corresponding primary antibodies. The membrane was then incubated with the appropriate horseradish peroxidase-conjugated secondary antibody, and the protein expression was detected using a SuperSignal West Dura substrate (Pierce). Each experiment was repeated three times and bands were quantified using Image-Pro Plus software.
Real-time PCR
LX-2 cells were plated in 6-well plates and cultured for 24 h. Cells at 70% confluence were treated with Withagulatin A (0−2 μmol/L) and 2 ng/mL TGF-β1 in serum free DMEM supplemented with 0.2% BSA for 24 h. Total RNA was extracted with TRIzol reagent (Generay Biotech, Shanghai, China) and then reverse transcribed to cDNA using a PrimeScriptTM RT reagent Kit (TaKaRa, Japan). Real-time PCR was performed using SYBR Green Realtime PCR Master Mix (TOYOBO, Japan) on a DNA Engine Opticon 2 System (Bio-Rad Laboratories, USA). The primer pairs were as follows: rat α1(I) procollagen: (F) 5′-CAC TCA GCC CTC TGT GCC-3′ and (R) 5′-ACC TTC GCT TCC ATA CTC G-3′; rat α2(I) procollagen: (F) 5′-AGA ATT CCG TGT GGA GGT TG-3′ and (R) 5′-GAG GGA GGG GAC TTA TCT GG-3′. The primer pairs for human α1(I) procollagen, α2(I) procollagen and rat 18s RNA were designed as previously described20, 22. The PCR cycle conditions were 95 °C for 10 s, 58 °C for 45 s and 72 °C for 30 s.
Smad2 nuclear translocation assay
The Smad2 nuclear translocation assay was performed using a CHO/EGFP-Smad2 stable cell line. Briefly, CHO/EGFP-Smad2 cells were seeded on 96-well plates and cultured for 24 h. The cells were pretreated with Withagulatin A (0-2 μmol/L) for 10 h and then treated with TGF-β (2 ng/mL) for 2 h. Finally, cell nuclei were stained with 2 μmol/L Hoechst 33342 for 15 min. Fluorescence images were taken by an INCell Analyzer 1000 (GE Healthcare) using the fluorescence microscope module and data were analyzed with the INCell Analyzer analysis software. Each treatment was repeated in 3 wells and pictures of 5 fields were taken for each well. The ratio of fluorescence intensity in the nucleus and cytoplasm was calculated by the software and defined as Nuc/Cyto Smad2 to indicate nuclear localization of Smad2. The experiment was repeated at least three times.
Statistical analysis
Results are expressed as means±SD. The statistical significant difference between treatment and control values was analyzed using one way ANOVA, which was followed by Dunnett's post test. A P value of less than 0.05 was considered statistically significant.
Results
Withagulatin A suppressed HSC activation and viability
Since activation and proliferation of hepatic stellate cells play pivotal roles in the progression of liver fibrosis, we initially evaluated the effects of Withagulatin A on α-SMA (a marker for HSC activation) expression and HSC viability. As expected, Withagulatin A treatment dose-dependently reduced α-SMA levels in activated HSCs (Figure 1B and 1C). In addition, we also investigated the effects of Withagulatin A on HSC viability using a SRB assay. These results demonstrated that Withagulatin A treatment for 48 h (as well as 24 and 72 h, data not shown) dramatically reduced rat primary HSC viability (P<0.01) in a dose-dependent manner (Figure 1D).
To further determine the inhibitory role of Withagulatin A on hepatic stellate cells in vitro, we studied its effect on the human hepatic stellate cell line LX-2. Similar to the results for primary HSCs, Withagulatin A treatment dose-dependently decreased α-SMA levels in LX-2 cells (Figure 1B and 1C). Additionally, Withagulatin A dramatically (P<0.05−0.001) reduced LX-2 viability in a concentration dependent manner (Figure 1D). In contrast, normal human hepatocytes (LO2 cells) were found to be resistant to Withagulatin A, and a marked effect on cell death (P<0.05) was only observed at the maximum concentration (20 μmol/L) after 48 h of treatment (Figure 1D). Similar results were obtained for 24 and 72 h of treatment (data not shown). We found that Withagulatin A at a concentration higher than 2 μmol/L caused a significant loss of cell viability. Therefore, in subsequent experiments we examined the mechanism underlying the actions of Withagulatin A with concentrations up to 2 μmol/L.
Withagulatin A induced S phase arrest in HSC
To explore the possible mechanism underlying the anti-proliferation activity of Withagulatin A, cell cycle progression in LX-2 cells was determined using flow cytometry. As indicated in Figure 2A, Withagulatin A treatment (0-2 μmol/L, 24 h) caused a dose-dependent increase in the number of cells in S phase and a corresponding decrease in the number of cells in G1 phase compared with DMSO treated control cells. Because the majority of cells were in the G0–G1 phase under normal conditions, we did not synchronize cells before flow cytometry.

Withagulatin A induces cell cycle arrest. (A) Withagulatin A arrests cells at S phase. LX-2 cells were incubated with Withagulatin A (0, 1, and 2 μmol/L) for 24 h. Cells were fixed, stained with propidium iodide, and analyzed by flow cytometry as described in Materials and methods. The experiment was repeated three times and representative data are shown. (B) Withagulatin A affects cell cycle regulatory proteins. LX-2 cells were incubated with Withagulatin A (2 μmol/L) for the indicated time points (0–24 h). Cells were harvested and cell lysates were analyzed by Western blotting for cyclin D1, p-CDK2, CDK2, and cyclin B1 levels. GAPDH was used as a loading control. Cells treated with DMSO were used as controls for each time point. Results shown are representative of three independent experiments. (C) Bands were quantified and data are expressed as fold of control. bP<0.05, cP<0.01 vs control cells.
We next assessed the influence of Withagulatin A on cell cycle regulatory proteins (Figure 2B and 2C). Cyclin D1, which controls G1-S transition, was initially induced by Withagulatin A treatment and was subsequently decreased after 12 h of treatment. In addition, the phosphorylation of CDK2, a G1 phase regulatory kinase, was regulated by Withagulatin A in a similar manner, which occurred without changes in total CDK2 protein levels. In contrast, the G2 phase cyclin, Cyclin B1, was decreased after Withagulatin A treatment. Thus, these results suggest that Withagulatin A may induce S phase arrest through dynamic regulation of Cyclin D1, CDK2, and Cyclin B1.
Withagulatin A induced apoptosis in HSCs
To determine whether the viability loss in Withagulatin A treated LX-2 cells could be attributed to the induction of apoptosis, we examined the morphological changes in Withagulatin A treated LX-2 cells with Hoechst 33342 staining. As shown in Figure 3A, the control cells showed round and homogeneously stained nuclei, whereas Withagulatin A treatment dose-dependently induced nuclear condensation and fragmentation in LX-2 cells (as indicated by arrows).

Withagulatin A induces cell apoptosis. (A) Induction of an apoptotic morphology by Withagulatin A. LX-2 cells were treated with Withagulatin A (0, 1, and 2 μmol/L) for 24 h. Cells were fixed, stained with Hoechst 33342, and visualized with a fluorescence microscope (scale bar=0 μm). Nuclear condensation and fragmentation are indicated by arrows. Representative views from five fields for each slide are shown and the treatments were performed in triplicate. (B) Withagulatin A affects apoptotic protein levels. LX-2 cells were treated with Withagulatin A (0, 1, and 2 μmol/L) for 24 h. Cells were harvested and lysates were analyzed by Western blot analysis to determine the levels of Bcl-2, Bax, cleaved-caspase3 (c-caspase3), and caspase3. GAPDH was used as a loading control. Results shown are representative of three independent experiments. (C) Bands were quantified and data are expressed as fold of control. bP<0.05, cP<0.01 vs control cells.
The expression of apoptotic proteins was examined to investigate the possible mechanism of Withagulatin A induced cell apoptosis after 24 h of treatment (Figure 3B and 3C), when maximum apoptosis was induced, but with limited cell death. We determined the protein levels of Bcl-2 and Bax, which have been reported to mediate cell apoptosis mainly through the mitochondrial pathway23. Withagulatin A treatment dose-dependently decreased the anti-apoptotic protein Bcl-2 while increasing the pro-apoptotic protein Bax. It has been reported that mitochondrial release of cytochrome c activates caspase3, which cleaves poly (ADP-ribose) polymerase (PARP) and other cellular targets that eventually lead to apoptosis24. We found that cleaved caspase3 was dose-dependently increased with a corresponding decrease of full length caspase3 levels after Withagulatin A treatment. Thus, these results suggested that Withagulatin A affected the mitochondrial pathway and subsequent targets such as the caspase cascade, which eventually induced apoptosis in LX-2 cells.
Withagulatin A suppressed Akt pathway
In order to further explore the underlying mechanism for the anti-fibrotic activity of Withagulatin A, we examined the effect of Withagulatin A on the Akt pathway, which promotes the cellular proliferation of HSCs25. As shown in Figure 4A and 4B, Withagulatin A treatment for 24 h dose-dependently decreased Akt phosphorylation at serine 473, whereas the total protein level was unaffected. The proteins downstream of Akt were also examined. Withagulatin A suppressed mTOR phosphorylation at serine 2448, which has been reported to be an Akt phosphorylation site8. Besides, Withagulatin A also repressed the phosphorylation of the mTOR substrate p70S6K at threonine 421 and serine 424, which has been shown to activate p70S6K26. In addition, Withagulatin A also induced a time-dependent decrease in AKT/mTOR/p70S6K phosphorylation as indicated in Figure 4C and 4D. As a positive control, the PI3K inhibitor LY294002 was used to inhibit AKT phosphorylation (Figure S3A).

Withagulatin A inhibits the Akt pathway. (A, B) Primary rat HSCs were incubated with Withagulatin A (0, 1, and 2 μmol/L) for 24 h (C, D) or treated with 2 μmol/L Withagulatin A for 0 to 24 h. Cells were harvested and analyzed by Western blotting for phosphorylated Akt (Ser 473), Akt, phosphorylated mTOR (Ser 2448), phosphorylated p70S6K (Thr 421/Ser 424) and p70S6K. GAPDH was used as a loading control. Results shown are representative of three independent experiments. Bands were quantified and data are expressed as fold of control. bP<0.05, cP<0.01 vs control cells.
Withagulatin A inhibited type I procollagen gene expression and Smad2/3 phosphorylation
The effect of Withagulatin A on ECM production was evaluated by real-time PCR analysis of α1/α2 procollagen I mRNA levels in primary HSCs (Figure 5A). Consistent with previous reports11, α1 and α2 procollagen I levels were significantly (P<0.001) up-regulated by TGF-β stimulation compared with the control group. Notably, Withagulatin A treatment significantly decreased TGF-β stimulated increases in type I procollagen mRNA expression (P<0.001) in comparison with the TGF-β treated group. Similar to rat primary hepatic stellate cells, LX-2 cells expressed high levels of ECM component proteins (including procollagen I) upon TGF-β stimulation. Our results demonstrated that Withagulatin A exhibited a similar inhibitory effect on procollagen I expression in LX-2 cells compared to primary HSCs (Figure 5B).

Withagulatin A inhibits TGF-β stimulated procollagen I mRNA expression and Smad2/3 phosphorylation. (A, B) Withagulatin A inhibits TGF-β stimulated procollagen I mRNA expression. (A) Primary rat HSCs and (B) LX-2 cells were treated with Withagulatin A (0, 1, and 2 μmol/L) in the presence of TGF-β (2 ng/mL) for 24 h in DMEM supplemented with 0.2% BSA. mRNA levels of α1 procollagen I and α2 procollagen I were analyzed by real-time PCR assays. Ribosomal 18s RNA was used as an internal control. bP<0.05, cP<0.01 compared with DMSO treated cells. (C) Withagulatin A suppresses TGF-β stimulated Smad2/3 phosphorylation. Primary rat HSCs were treated with Withagulatin A (0, 1, and 2 μmol/L) in the presence of TGF-β (2 ng/mL) for 24 h in DMEM supplemented with 0.2% BSA. Cells were harvested and subjected to Western blot analysis for phosphorylated Smad2 (Ser 465/467), Smad2, phosphorylated Smad3 (Ser 432/425), and Smad3. GAPDH was used as a loading control. Results shown are representative of three independent experiments. (D) Bands were quantified and data are expressed as fold of control. (E, F) Withagulatin A suppresses TGF-β induced Smad2 nuclear translocation. CHO/EGFP-Smad2 cells were pretreated with Withagulatin A (0, 1 and 2 μmol/L) for 10 h and then stimulated with TGF-β (2 ng/mL) for 2 h in serum free F-12 medium supplemented with 0.2% BSA. Finally, cells were stained with 2 μmol/L Hoechst 33342 for 15 min and fluorescent images were taken by an INCell Analyzer 1000. Each treatment was repeated in 3 wells and 5 fields were photographed for each well. (E) Representative views are presented (scale bar=50 μm) and (F) data were quantified using the INCell Analyzer analysis software. cP<0.01 vs TGF-β stimulated cells.
The TGF-β/Smad pathway plays a pivotal role in TGF-β stimulated collagen gene expression. Therefore, to investigate the mechanism underlying the inhibition of TGF-β stimulated type I procollagen gene expression by Withagulatin A, we examined the phosphorylation status of Smad proteins. We found that TGF-β induced a maximal effect at 2 ng/mL for 2 h (data not shown). Therefore, we used this condition for the following experiments. As shown in Figure 5C and 5D, TGF-β potently induced Smad2 and Smad3 phosphorylation, whereas Withagulatin A treatment dramatically inhibited TGF-β induced Smad2 or Smad3 phosphorylation but did not affect either of their protein levels. As a positive control, the TGF-βR1 inhibitor SB431542 also blocked TGF-β induced Smad3 phosphorylation (Figure S3B).
Phosphorylated Smad proteins translocate to the nucleus and promote collagen gene transcription. Therefore, we assessed the effect of Withagulatin A on TGF-β stimulated Smad2 translocation. As shown in Figure 5E, the majority of EGFP-Smad2 protein was localized to the cytoplasm in unstimulated cells and translocated to the nucleus upon TGF-β stimulation. However, Withagulatin A dose-dependently blocked TGF-β stimulated Smad2 nuclear translocation as quantified in Figure 5F (P<0.001). In addition, to test whether Withagulatin A could affect EGFP translocation, we used the U2OS-EGFP-Rev cell line (Bioimage, Thermol). In this cell line, EGFP has been fused to the HIV-1 regulatory protein Rev, which contains a nuclear export signal and a nuclear localization signal in its carboxy- and amino-termini, respectively. Treatment of U2OS-EGFP-Rev cells with Withagulatin A did not induce nuclear translocation of EGFP (Figure S2), thereby indicating that Withagulatin A did not affect EGFP translocation.
Discussion
Proliferation and ECM production of activated hepatic stellate cells constitute the major pathways contributing to hepatic fibrosis3. Therefore, inhibition of the accumulation of activated HSCs and the prevention of extracellular matrix deposition are currently considered promising anti-fibrotic strategies2. We report here that Withagulatin A, which was extracted from Physalis angulata L, inhibited hepatic stellate cell proliferation and type I procollagen expression by targeting the Akt/mTOR/p70S6K and TGF-β/Smad pathways (Figure 6).

Proposed model illustrating the anti-fibrotic mechanism of Withagulatin A (With A). During liver fibrosis, stimulatory signals from the fibrogenic cytokines including PDGF and TGF-β are transduced into target cells through their corresponding receptors, which in turn activate Akt and Smad proteins by phosphorylation. Withagulatin A inhibited the phosphorylation of Akt and its downstream targets such as mTOR and p70S6K, which finally led to reduced HSC proliferation. Meanwhile, Withagulatin A suppressed Smad2/3 phosphorylation and consequently blocked translocation to the nucleus, thereby resulting in reduced transcription of ECM proteins.
Rodent primary HSCs27 and a human hepatic stellate cell line20, which retain key features of primary HSCs, have been adopted as useful tools for liver fibrosis research. Hepatic stellate cells respond to PDGF during their activation process and produce PDGF through autocrine pathways. Both primary HSCs and the LX-2 cell line are activated hepatic stellate cells; therefore, they were both stimulated by autocrine cytokines. Furthermore, the inhibitory effects of Withagulatin A on fully activated HSCs might indicate therapeutic opportunities for treating the late stages of liver fibrosis. To test the possible cytotoxicity of Withagulatin A, we used the normal human hepatocyte cell line LO2. Withagulatin A did not obviously decrease cell viability of LO2 cells even at high concentrations when compared with HSCs. Nevertheless, further studies are required to evaluate the toxicity of Withagulatin A on other cell lines and in in vivo experiments. Moreover, the fact that Withagulatin A inhibited production of the activation marker α-SMA implies that this natural product might also exhibit inhibitory activity against HSC activation. However, further investigations are required to examine this effect due to the complicated regulatory mechanisms underlying HSC activation and α-SMA expression. Nevertheless, down-regulation of α-SMA levels in activated HSCs implied the possibility of fibrotic reversion, which would be of great importance but difficult to achieve28.
Our results suggested that Withagulatin A inhibited HSC proliferation by arresting cells in the S phase. Dynamic fluctuations of cyclin levels among the different cell cycle stages regulate the activity of these kinase complexes, which subsequently control cell cycle progression. Cyclin D1 and CDK2 are essential regulators for G1-S transition29, 30, and Cyclin B1 regulates cell entry into mitosis31. Withagulatin A treatment initially increased Cyclin D1 levels and CDK2 phosphorylation, but subsequently decreased them, which was in concordance with the flow cytometry data that indicated an accumulation of cells in the S phase and a decrease in the number of cells in the G1 phase. In addition, Withagulatin A treatment time-dependently decreased Cyclin B1 levels and consequently hindered cell entry into the G2/M phase. Inhibition of proliferation and induction of apoptosis in HSCs are both effective means to clear activated HSCs32. Our work demonstrated that in addition to the inhibition of HSC proliferation, Withagulatin A also induced LX-2 cell apoptosis, as indicated by the nuclear condensation and induction of apoptotic regulatory proteins. However, due to the tightly adhesive nature of HSCs, we were unable to perform annexin V staining for apoptosis.
The inhibition of PI3K activity has been shown to suppress cell proliferation in activated HSCs25. Meanwhile, p70S6K regulates protein synthesis and proliferation, and the blocking of mTOR (its upstream kinase) has been shown to inhibit DNA synthesis and Cyclin D1 expression in HSCs10. Our results showed that Withagulatin A decreased Akt, mTOR, and p70S6K phosphorylation levels, thereby indicating that the Akt/mTOR/p70S6K signaling pathway may at least be partially accountable for the inhibitory effect of Withagulatin A on HSC proliferation.
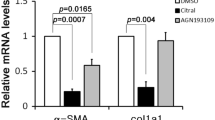
Type I collagen is the major ECM component in fibrotic liver32 and is potently stimulated by the fibrogenic cytokine TGF-β. Withagulatin A reduced TGF-β stimulated α1 and α2 procollagen I mRNA expression in both rat primary HSCs and LX-2 human hepatic stellate cells. However, poor antibody availability made it hard to detect collagen protein levels. Smad proteins have been reported to transduce TGF-β signaling to the nucleus and regulate collagen gene expression11. Our results demonstrated that Withagulatin A inhibited TGF-β stimulated Smad2 and Smad3 phosphorylation and further blocked TGF-β induced Smad2 nuclear translocation. In addition, Withagulatin A also inhibited basal procollagen I mRNA expression but not basal Smad2 or Smad3 phosphorylation (Figure S1), which may have been due to the different regulatory mechanisms between basal and TGF-β stimulated procollagen I transcription33.
In summary, the current work indicated that Withagulatin A from Physalis angulata L inhibited hepatic stellate cell proliferation and type I procollagen expression. These findings suggest that this natural product may assist in the development of anti-fibrogenic reagents for future studies.
Author contribution
Qiong LIU designed and performed the research and wrote the paper; Xu WANG helped with primary cell cultures; Jing CHEN and Liang YU helped in providing reagents; Li-hong HU provided the Withagulatin A compound; Xu SHEN critically revised the paper.
References
Albanis E, Friedman SL . Hepatic fibrosis. Pathogenesis and principles of therapy. Clin Liver Dis 2001; 5: 315–34, v–vi.
Bataller R, Brenner DA . Liver fibrosis. J Clin Invest 2005; 115: 209–18.
Friedman SL . Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 2000; 275: 2247–50.
Geerts A, Lazou JM, De Bleser P, Wisse E . Tissue distribution, quantitation and proliferation kinetics of fat-storing cells in carbon tetrachloride-injured rat liver. Hepatology 1991; 13: 1193–202.
Mak KM, Leo MA, Lieber CS . Alcoholic liver injury in baboons: transformation of lipocytes to transitional cells. Gastroenterology 1984; 87: 188–200.
Scheid MP, Woodgett JR . PKB/AKT: functional insights from genetic models. Nat Rev Mol Cell Biol 2001; 2: 760–8.
Marra F, Gentilini A, Pinzani M, Choudhury GG, Parola M, Herbst H, et al. Phosphatidylinositol 3-kinase is required for platelet-derived growth factor's actions on hepatic stellate cells. Gastroenterology 1997; 112: 1297–306.
Memmott RM, Dennis PA . Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal 2009; 21: 656–64.
Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 2006; 22: 159–68.
Gabele E, Reif S, Tsukada S, Bataller R, Yata Y, Morris T, et al. The role of p70S6K in hepatic stellate cell collagen gene expression and cell proliferation. J Biol Chem 2005; 280: 13374–82.
Inagaki Y, Okazaki I . Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut 2007; 56: 284–92.
Abdollah S, Macias-Silva M, Tsukazaki T, Hayashi H, Attisano L, Wrana JL . TbetaRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J Biol Chem 1997; 272: 27678–85.
Lotersztajn S, Julien B, Teixeira-Clerc F, Grenard P, Mallat A . Hepatic fibrosis: molecular mechanisms and drug targets. Annu Rev Pharmacol Toxicol 2005; 45: 605–28.
Lin YS, Chiang HC, Kan WS, Hone E, Shih SJ, Won MH . Immunomodulatory activity of various fractions derived from Physalis angulata L extract. Am J Chin Med 1992; 20: 233–43.
Wu SJ, Ng LT, Chen CH, Lin DL, Wang SS, Lin CC . Antihepatoma activity of Physalis angulata and P peruviana extracts and their effects on apoptosis in human Hep G2 cells. Life Sci 2004; 74: 2061–73.
Glotter E . Withanolides and related ergostane-type steroids. Nat Prod Rep 1991; 8: 415–40.
Chen CM, Chen ZT, Hsieh CH, Li WS, Wen SY . Withangulatin A, a new Withanolide from Physalis angulata. Heterocycles 1990; 31: 1371–5.
He QP, Ma L, Luo JY, He FY, Lou LG, Hu LH . Cytotoxic withanolides from Physalis angulata L. Chem Biodivers 2007; 4: 443–9.
Siegmund SV, Uchinami H, Osawa Y, Brenner DA, Schwabe RF . Anandamide induces necrosis in primary hepatic stellate cells. Hepatology 2005; 41: 1085–95.
Xu L, Hui AY, Albanis E, Arthur MJ, O'Byrne SM, Blaner WS, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut 2005; 54: 142–51.
Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990; 82: 1107–12.
Jinnin M, Ihn H, Tamaki K . Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-beta1-induced extracellular matrix expression. Mol Pharmacol 2006; 69: 597–607.
Chao DT, Korsmeyer SJ . BCL-2 family: regulators of cell death. Annu Rev Immunol 1998; 16: 395–419.
Thornberry NA, Lazebnik Y . Caspases: enemies within. Science 1998; 281: 1312–6.
Reif S, Lang A, Lindquist JN, Yata Y, Gabele E, Scanga A, et al. The role of focal adhesion kinase-phosphatidylinositol 3-kinase-akt signaling in hepatic stellate cell proliferation and type I collagen expression. J Biol Chem 2003; 278: 8083–90.
Pullen N, Thomas G . The modular phosphorylation and activation of p70s6k. FEBS Lett 1997; 410: 78–82.
Friedman SL, Roll FJ, Boyles J, Arenson DM, Bissell DM . Maintenance of differentiated phenotype of cultured rat hepatic lipocytes by basement membrane matrix. J Biol Chem 1989; 264: 10756–62.
Friedman SL, Bansal MB . Reversal of hepatic fibrosis — fact or fantasy? Hepatology 2006; 43: S82–8.
Stillman B . Cell cycle control of DNA replication. Science 1996; 274: 1659–64.
Morgan DO . Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 1997; 13: 261–91.
Norbury C, Nurse P . Animal cell cycles and their control. Annu Rev Biochem 1992; 61: 441–70.
Friedman SL . Mechanisms of hepatic fibrogenesis. Gastroenterology 2008; 134: 1655–69.
Cutroneo KR . How is Type I procollagen synthesis regulated at the gene level during tissue fibrosis. J Cell Biochem 2003; 90: 1–5.
Acknowledgements
We are grateful to Dr SL FRIEDMAN (Mount Sinai School of Medicine, New York) for kindly providing the human stellate cell line LX-2.
This work was supported by the National Natural Science Foundation of China (grants 20721003, 90713046), the State Key Program of Basic Research of China (grants 2009CB918502, 2010CB912501), the Shanghai Basic Research Project (09QA1406800), the Foundation of Chinese Academy of Sciences (KSCX1-YW-02-2) and the Key New Drug Creation and Manufacturing Program (2009ZX09301-001).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary information is available at Acta Pharmacologica Sinica's website.
Supplementary information
Supplementary information
Effect of Withagulatin A on procollagen I expression and Smad2/3 phosphorylation. (DOC 454 kb)
Rights and permissions
About this article
Cite this article
Liu, Q., Chen, J., Wang, X. et al. Withagulatin A inhibits hepatic stellate cell viability and procollagen I production through Akt and Smad signaling pathways. Acta Pharmacol Sin 31, 944–952 (2010). https://doi.org/10.1038/aps.2010.72
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2010.72
