
ABSTRACT
Interleukin (IL)-8 is a potent neutrophil chemotactic factor and a crucial mediator in neutrophil-dependent inflammation. Various cell types produce IL-8, either in response to external stimuli such as cytokines or bacterial infection, or after malignant transformation. Anti-IL-8 strategies have been considered for anti-inflammatory therapy. In this paper we demonstrate that the RNA interference technique can be used to efficiently down-regulate IL-8 protein expression in airway epithelial cells. We used a helper-dependent adenoviral vector to express a small hairpin (sh)RNA targeting human IL-8 in cultured airway epithelial cells (IB3-1, Cftr−/−; C38, Cftr-corrected) stimulated with TNF-α, IL-1β or heat-inactivated Burkholderia cenocepacia. Stimulated IL-8 expression in IB3-1 and C38 cells was significantly reduced by shRNA expression. The shRNA targeting IL-8 had no effect on the activation of NF-κB, or on the protein levels of IκB or IL-6, suggesting that this anti-IL-8 strategy was highly specific, and therefore may offer potential for the treatment of inflammatory diseases.
Similar content being viewed by others


INTRODUCTION
IL-8 is a potent neutrophil chemoattractant and activator 1, and high levels of IL-8 are frequently found to be associated with a variety of inflammatory diseases with neutrophil infiltration. For example, in airways of cystic fibrosis patients, the production of IL-8 is exaggerated, leading to excessive neutrophil infiltration 2. Patients with ulcerative colitis display increased levels of IL-8 in inflamed areas of the gut 3. In addition, IL-8 is induced in synovial cells of rheumatoid arthritis patients 4, and in the myocardium after ischemia and reperfusion in vivo 5. Interestingly, elevated levels of IL-8 in the sputum of cystic fibrosis patients is reduced by either antibiotic therapy or gene therapy with AAVCFTR 6, 7. In certain gene therapy applications inflammation due to the therapy is a problem 8, and addition of an anti-IL-8 treatment may be helpful.
IL-8, also called CXCL8, belongs to the CXC chemokine family 9. IL-8 expression is tightly regulated and requires activation of NF-κB as well as MAP kinases 10, 11. Certain cytokines, such as TNF-α and IL-1β, and many microbial products including lipopolysaccharide, tri-acyl lipopeptides and bacterial flagellin, are potent inducers of IL-8 expression in a variety of cells, such as macrophages and epithelial cells. Induction of IL-8 by TNF-α or IL-1β is mediated through their corresponding receptors, which activate the NF-κB signaling pathway 12, 13. Similarly, microbial products induce IL-8 expression via a family of plasma membrane receptors called Toll-like receptors (TLR) 14, 15, 16. TLRs are expressed in many types of cells including airway and intestinal epithelial cells 17. They are part of the innate immune system for activating the expression of IL-8 and other genes to fight infection.
Anti-IL-8 antibodies have been shown to reduce lung reperfusion injury in rabbits 18, suggesting that an anti-IL-8 strategy may be used to control tissue inflammation and aid disease management. Antibodies against IL-8 are effective for reduction of IL-8 activity in animal studies 19, 20, but it is not clear whether this approach would work in humans. Since administration of antibodies is a systemic approach, this may compromise the host's ability to fight infection. In addition, repeated administration of antibodies can be very expensive. An alternative approach is to use CXCR2 receptor antagonists to block neutrophil influx and reduce inflammation. It has been shown that SB-265610, a non-peptide CXCR2 antagonist, can prevent neutrophil accumulation in hyperoxia-exposed newborn rats 21.
In this paper, we report a new tool for down-regulating IL-8 expression by using RNA interference (RNAi). RNAi is a relatively new discovery in post-transcriptional gene silencing 22 and an important tool for gene function analysis 23, 24, 25, 26. The term RNAi was initially coined by Fire and coworkers 27 to describe the observation that double-stranded RNA targeted to a gene of interest, when introduced into nematodes, can result in degradation of the corresponding mRNA. This powerful technique was then developed for mammalian cells 23. Since synthetic RNA oligos are not cost-effective, several groups have successfully developed DNA-based vectors to express short double-stranded RNA with a small hairpin loop structure (shRNA) to serve as small interfernce (si)RNA 25, 26, 28, 29. We have designed an expression cassette using the U6 promoter to drive a shRNA targeting the IL-8 mRNA. Since the efficiency of gene transfer and episomal stability of adenoviral vectors is far superior to plasmids, we delivered the shRNA expression cassette to cultured human airway epithelial cells using a helper-dependent adenovirus vector (HD-Ad). HD-Ads 30 were developed in gene therapy research to overcome strong host immune responses to conventional adenoviral vectors. In HD-Ads, all adenoviral sequences are deleted except the viral inverted terminal repeats located at each end of the genome, and the viral packaging signal. The deletion of the viral genes prolongs transgene expression and also increases the cloning capacity to 36 kb 30. We show that an shRNA expressed from an HD-Ad can effectively down-regulate endogenous IL-8 expression induced by proinflammatory cytokines or bacteria, in Cftr−/− and Cftr-corrected lung epithelial cells.
Materials and methods
Cell culture
IB3-1 (ΔF508/w1282X) and C38 cells (CFTR-corrected) were described in 31, 32. IB3-1 and C38 cells were cultured in LHC-8 medium containing 5% FBS and antibiotics (2.5 μg/ml fungizone, 80 μg/ml tobramycin, 0.2mg/ml primaxin and 100U/ml each of penicillin and streptomycin). COS-1 cells were grown in DMEM with10% FBS.
IL-8 RNAi design
An siRNAi-expressing construct was designed as described 25, using a mouse U6 gene promoter to drive expression. A target region of IL-8 was selected beginning with three guanines (5′-GGG TGC AGA GGG TTG TGG AGA-3′). Shown in Fig. 1A, top, two sets of DNA oligonucleotides were used to construct the coding region for the short double-stranded RNA with a hairpin. A HindIII site was designed at the hairpin to facilitate restriction analysis for the cloning. In the second pair of oligonucleotides, the sequence of the top strand ends with TTTTT, which serves as Pol III transcription termination signal. The plasmid pBSII-SK (Stratagene, La Jolla, CA) was used as the backbone for the final plasmid, named pBS/U6-IL8. All DNA oligonucleotides were purchased from ACTG Inc. (Toronto, ON, Canada).

Design of an siRNA construct targeting IL-8 and functional assay with a reporter system. (A) siRNA expression plasmid and the reporter system. The top part shows a strategy for generating siRNA from a DNA template in vivo. Two pairs of oligonucleotides are linked by Hind III site and inserted after the U6 promoter. The resulting RNA is predicated to fold back to form shRNA. The lower panel shows a reporter plasmid containing a target sequence from IL-8. The target sequence was inserted between the CMV promoter and SEAP gene before the start codon. (B) Analysis of the effectiveness of the IL-8 shRNA with the reporter system in COS-1 cells. COS-1 cells were transfected with 0.05 μg of pCMVSEAP or pCMVIL-8SEAP target, together with 1.5 μg pBS/U6-IL-8RNAi or empty vector (pBS/U6). SEAP activity was analyzed 48 h post transfection. The average of three independent experiments is shown; error bars indicate mean ± SE. *P < 0.05, pIL-8CMVSEAP+vector versus pIL-8CMVSEAP+IL-8RNAi.
Reporter system for measuring gene knock-down
A reporter plasmid for measuring IL-8 knock-down was constructed by inserting a 21 bp double-stranded oligonucleotide containing the IL-8 targeted site into the pCMVSEAP reporter plasmid, between the CMV promoter and the translation initiation codon of the secreted alkaline phosphatase (SEAP) gene in pSEAP2 (Clontech, Palo Alto, CA) (Fig. 1A, bottom). The resulting plasmid, pCMV-IL-8SEAP, expresses the same level of SEAP activity as its parental plasmid, pCMVSEAP. COS-1 cells were cotransfected with pCMV-IL-8SEAP and pBS/U6IL-8RNAi in a ratio of 1:30 (by moles) using PolyFect Transfection Reagent (QIAGEN, Toronto, Canada). Cell culture media were collected for SEAP analysis 48 h following cotransfection, and SEAP assays were performed using the Phospha-Light System (Applied Biosystems, Bedford, MA).
HD-Ad-IL8RNAi production
A BssHII-DNA fragment containing the U6 promoter and a short sequence for IL-8 RNAi expression was excised from pBSII/U6/IL-8RNAi and ligated into the AscI site of pC4HSU (Merck and Co., USA) (Supplementary Fig. 1). The IL-8RNAi-C4HSU DNA was treated with PmeI to expose the viral ITRs and introduced into 293cre4 cells using the CellPhect Transfection kit (Amersham Pharmacia, Buckinghamshire, UK). The preparation and purification of viral vectors were carried out as described 33.
Viral transduction
To determine the number of virus particles per cell required to achieve transduction of ∼100% cells, we tested transduction frequency using 0, 25, 50 and 100 moi (number of infectious units per cell) of a lacZ-expressing HD-Ad. We found that 50 moi can achieve almost 100% transduction of IB3-1 and C38 cells.
Induction and ELISA
IB3-1 and C38 cells were treated with (or without) HD-AD-IL8RNAi for 4 h, then induced for 24 h with 5 ng/ml TNF-α, or 1 ng/ml IL-1β, or 50 cfu of Burkholderia cenocepacia (Bc; clinical isolate BC7, strain ET12) that was heat inactivated (20 min at 60°C). Human recombinant TNF-α and IL-1β were purchased from R&D Systems (Minneapolis, MN). Conditioned media were collected and stored at −80°C. IL-8 and IL-6 protein levels were measured by ELISA (Bender MedSystems, Diagnostics GmbH, Vienna). The statistical analysis was performed with the two-tailed t-test.
EMSA
Nuclear extracts were prepared as described 34. A DNA oligonucleotide probe containing the κ-light chain enhancer NF-κB consensus binding site (5′-AGTTGAGGGGACTTTCCCAGG C-3′) was end-labelled with γ-32P-ATP. Five micrograms of nuclear extracts were used for each reaction. Complexes were resolved on 6% non-denaturing polyacrylamide gels.
Western Blot analysis
Ten μl total cell lysate from each sample were run on 10% SDS-polyacrylamide gel and transferred to a nitrocellulose filter, then probed with rabbit polyclonal antibody against IκB-α/β (Santa Cruz Biotechnology, Santa Cruz, CA). Protein bands were visualized using alkaline phosphatase-conjugated anti-rabbit secondary antibody (Amersham, Bioscienes, UK).
RESULTS
Analysis of shRNA targeting efficiency using a reporter gene system
We built an shRNA expression plasmid, pBS/U6-IL-8RNAi, using an U6 promoter-based plasmid vector 25 to produce an shRNA targeting the human IL-8 mRNA (Fig. 1A, top). To determine the silencing efficiency of the shRNA, we designed a SEAP reporter plasmid with an IL-8 target sequence at the 5′ end of the SEAP coding region of pCMV-SEAP (Fig. 1A, lower). The level of SEAP expression from the target plasmid, pCMV-IL-8-SEAP, was comparable to that from the parental plasmid when co-transfected with the control plasmid pBS/U6 into COS-1 cells (Fig. 1B). To determine whether pBS/U6-IL-8RNAi was functional, we introduced it together with the reporter target plasmid (pCMV-IL-8-SEAP) into COS-1 cells. pBS/U6-IL-8RNAi significantly reduced SEAP production from pCMV-IL-8-SEAP (∼80%). pBS/U6-IL-8RNAi had no effect on SEAP expression from the parental plasmid (pCMV-SEAP) which does not contain the IL-8 target sequence. As reported in other studies 23, 25, our co-transfection analysis was done with a high ratio of RNAi/target plasmid (30:1). To enhance the tranduction efficiency and stability we decided to use an HD-Ad to deliver the IL-8 RNAi cassette.
IL8RNAi-HD-Ad reduces endogenous IL-8 expression induced by TNF-α or IL-1β
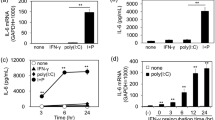
The reporter analysis indicated indirectly that the shRNA expressed from pBS/U6-IL8RNAi can knock down IL-8 mRNA. We next designed an HD-Ad vector (Supplementary Fig. 1) to express the shRNA targeting IL-8. We found that 50 moi (number of infectious unit per cell) was sufficient to achieve almost 100% cell transduction (data not shown). Four hours following treatment of IB3-1 or C38 cells with IL8-RNAi-HD-Ad, or the control virus HD-Ad-LacZ, the cells were cultured in the presence of TNF-α or IL-1β for an additional 24 h. The level of IL-8 in both cell lines was very low (undetectable in C38) under normal growth conditions, but was induced in the presence of TNF-α or IL-1β. Shown in Fig. 2A and B, the level of IL-8 protein was 3-4.5-fold higher in CF cells (IB3-1) than in Cftr-corrected cells (C38) when induced by TNF-α or IL-1β. IL-8 induction by TNF-α or IL-1β in each cell line was inhibited ∼70% when the cells were pre-treated with IL8RNAi-HD-Ad. Similar results were obtained following cytokine stimulation for 48 hours (data not shown). The control virus HD-Ad-lacZ did not induce expression of IL-8 (Fig. 2A, B), nor did it affect TNF-α or IL-1β induction of IL-8 in either cell line (data not shown).

Reduction of cytokine-induced IL-8 expression by shRNA expressed from a helper-dependent vector. IB3-1 cells (A) or C38 cells (B) were infected with IL-8RNAi-HD-Ad or HD-Ad-LacZ (as a viral vector control) and induced with 5 ng/ml TNF-α or 1 ng/ml IL-1β. IL-8 protein was analyzed by ELISA. The average of three independent experiments is shown; error bars indicate mean ± SE. *P < 0.05, TNF-α or IL-1β induced cells receiving IL-8RNAi-HD-Ad versus induced cells not receiving HD-Ad.
IL8RNAi-HD-Ad reduces endogenous IL-8 expression induced by heat-inactivated bacteria
Burkholderia cenocepacia (bacteria belonging to genomovar III of the Burkholderia cepacia complex) is an opportunistic pathogen found in patients with cystic fibrosis and induces IL-8 production in lung epithelial cells 11. We examined whether IL8RNAi-HD-Ad could knock down B. cenocepacia-induced IL-8 expression in airway epithelial cells. We heat-inactivated the bacteria so their numbers could be easily controlled in cell culture. Shown in Fig. 3, the level of IL-8 expression induced by heat-inactivated Bc was greatly exaggerated (6-fold) in Cftr−/− (IB3-1) cells compared to Cftr-corrected (C38) cells. The peak of IL-8 protein occurred 24 h following induction (data not shown). IL8RNAi-HD-Ad significantly reduced expression of endogenous IL-8 protein induced by Bc, in both cell lines, and the degree of reduction (64-70%) was similar to that observed in cells induced with TNF-α or IL-1β prior to the addition of IL8RNAi-HD-Ad (Fig. 2A, B).

Reduction of bacterium-induced IL-8 expression by shRNA expressed from a helper-dependent vector. IB3-1 or C38 cells transduced with IL-8RNAi-HD-Ad were stimulated with heat-inactivated B. cenocepacia (Bc) for 24 h. IL-8 protein was determined by ELISA. The average of three independent experiments is shown; error bars indicate mean ± SE. *P < 0.05 , cells receiving IL-8RNAi-HD-Ad versus those not receiving HD-Ad.
IL8RNAi-HD-Ad-mediated IL-8 reduction does not affect NF-κB activation.
Previous studies have demonstrated that TNF-α or IL-1β stimulate expression of IL-8 and IL-6 through activation of the NF-κB pathway 12, 35. Although the RNAi strategy targeting IL-8 is expected to be gene-specific, non-specific down-regulation of NF-κB activity could also reduce the level of IL-8 expression. To test this possibility, we performed EMSA to examine the NF-κB DNA-binding activity in nuclear extracts from cells receiving various treatments. IL-8RNAi-HD-Ad did not interfere with TNF-α or IL-1β induced NF-κB activation in IB3-1 or C38 cells (Fig. 4A, lanes 5, 7 and lanes 11, 13). There was no change in NF-κB DNA-binding activity in un-induced cells, regardless of whether they were first transduced with IL-8RNAi-HD-Ad (Fig. 4A, lanes 8 and 14). Similarly, IL-8RNAi-HD-Ad did not interfere with Bc-induced NF-κB activation (Fig. 4B). Under quiescent conditions NF-κB is inhibited by binding to IκB in the cytoplasm. Upon stimulation IκB is degraded, allowing NF-κB to translocate to the nucleus and modulate inflammatory gene expression 36. Both IκB-α and IκB-β were degraded quickly (in 15 min) following stimulation with TNF-α or IL-1β, regardless of whether the cells were first transduced with IL-8RNAi-HD-Ad. IL-8RNAi-HD-Ad had no effect on the levels of IκB-α and -β in uninduced cells (Fig. 5).


EMSA of NF-κB DNA-binding activity in IB3-1 and C38 cells treated with IL-8RNAi-HD-Ad and stimulated with TNF-α, IL-1β or Bc. IB3-1 or C38 cells with (or without) tranduction with IL-8RNAi-HD-Ad for 4 h were treated with TNF-α or IL-1β for 2 h (A), or with heat-inactivated Bc (50 cfu) for 4 h (B). Nuclear extracts were prepared for NF-κB DNA-binding activity assay. Specificity of complex binding was examined by competition with unlabeled (cold) and nonspecific (NS) probes (50 ng). Nuclear extracts from cells without stimulation or viral transduction were included as controls. * indicates a non-specific DNA protein complex.

IκB-α and IκB-β protein levels in IB3-1 and C38 cells after TNF-α or IL-1β induction. IB3-1 or C38 cells were transduced with IL-8RNAi-HD-Ad for 4 h and then treated with TNF-α or IL-1β for 15 min. Whole cell lysates were collected to detect IκB-α and -β protein by immunoblotting. A monoclonal antibody against β-actin was used as a protein loading control.
IL8RNAi-HD-Ad does not affect IL-6 induction
Expression of both IL-6 and IL-8 is induced by TNF-α or IL-1β through NF-κB activation 12. To examine whether IL-6 induction by TNF-α or IL-1β is non-specifically affected by IL-8RNAi, we measured IL-6 activity in IB3-1 and C38 cells transduced with IL-8RNAi-HD-Ad. Shown in Fig. 6, there was a very low level of IL-6 protein secreted from the unstimulated cells. IL-6 protein was greatly induced by incubation with TNF-α, IL-1β or Bc for 24 h. However, there was no difference in expression of IL-6 between cells treated with or without IL-8RNAi-HD-Ad. In contrast to IL-8, there was little difference in IL-6 induction between CF and Cftr-corrected cells.

Effect of IL-8RNAi on IL-6 expression induced with TNF-α or IL-1β. IB3-1 (A) and C38 (B) cells were transduced with IL-8RNAi-HD-Ad or HD-Ad-lacZ (as a viral vector control). IL-6 protein expression was induced with 5 ng/ml of TNF-α, 1 ng/ml IL-1β or Bc. IL-6 protein expression was analyzed by ELISA. The average of three independent experiments is shown; error bars indicate mean ± SE.
DISCUSSION
Induction of IL-8 occurs in a variety of inflammatory diseases, including cystic fibrosis, chronic obstructive pulmonary disease (COPD), acute respiratory distress syndrome (ARDS), asthma, idiopathic pulmonary fibrosis, meconium aspiration syndrome, bacterial pneumonia, psoriasis, rheumatiod arthritis, inflammatory bowel disease and septic shock 10, 37, 38, 39, 40, 41. Boekholdt et al demonstrated that among apparently healthy men and women, elevated levels of IL-8 are associated with an increased risk of future coronary artery disease 42. Recent reports also indicate that IL-8 plays an important role in atherosclerosis, and in mediating angiogenic activity in tumor growth and metastasis 19, 43, 44.
Strategies for inhibiting IL-8 include neutralizing the protein with fully humanized antibodies against IL-8 45, 46. In a rabbit model of lung reperfusion injury, reperfusion of the ischaemic lung induced local IL-8 production, massive neutrophil infiltration and destruction of pulmonary tissue. Administration of a neutralizing monoclonal antibody against IL-8 prevented neutrophil infiltration and tissue injury 18. IL-8 antibody administration also significantly inhibited proteinuria, as well as infiltration of neutrophils into rabbit glomeruli, in an acute glomerulonephritis animal model initiated by deposition of immune complex 20. In a mouse orthotopic bladder cancer model, a significant decrease in human tumor growth and invasion was observed after IL-8 antibody treatment 19, 47. These observations suggest that IL-8 could be a target for therapeutic intervention in neutrophil-mediated acute inflammation, and possibly carcinogenesis. But the use of humanized antibodies in clinical applications is hindered by a combination of factors including a brief half-life and fast blood clearance, their immunogenicity, and high cost.
Recent advances in sequence-specific gene silencing by double-stranded RNAs (RNA interference) provides a new platform to develop gene-specific drugs for disease management. Our results indicated that an IL-8RNAi expression plasmid significantly reduced the activity of a reporter gene (SEAP) containing an IL-8 target sequence. Although the use of plasmid-based shRNA expression has quickly become the preferred method for targeted gene silencing, there are limitations. The delivery of plasmids to certain types of cells such as primary cells, neural cells and stem cells is hampered by poor transfection efficiency. Delivery of expression plasmids to mammals as gene therapy is very inefficient, particularly to the lung 48. The delivery of shRNA by viral transduction is one way to overcome these shortcomings. Some groups 49, 50, 51, 52 have reported that efficient introduction and stable integration of shRNA-expression cassettes can be achieved using Moloney murine leukaemia virus, lentiviral vector systems or the first-generation adenoviral vectors. However, the first-generation Ad vector is not a good choice for delivery of anti-inflammatory shRNAs to mammals, since the vector itself provokes strong host inflammatory responses. HD-Ads cause significantly less inflammation in mammals since they do not express adenoviral genes, although some inflammation due to the capsid proteins still occurs. Like earlier generations of Ads, HD-Ads transduce most cell types very effectively. In addition, the integration frequency of vector DNA into the host genome is very low 53, thus reducing risk of insertional mutagenesis in clinical applications. One concern about using HD-Ads, versus first-generation Ads, is the technical difficulty in producing these vectors. However, Palmer and Ng reported a new method that can shorten the production time of HD-Ads to 12 days, and produce >1013 viral particles 54.
Microbial products can activate the NF-κB signaling pathway in airway epithelial cells 55, 56. This activation is initiated through interactions between microbial products and various Toll-like receptors present in airway epithelial cells 57. Activation of the NF-κB pathway via TLRs can induce the expression of cytokines, including IL-8, which leads to inflammation. Furthermore, since IL-8 is a potent chemoattractant and activator of neutrophils, overexpression of IL-8 can lead to excessive neutrophil infiltration and tissue damage. The level of IL-8 in CF airway is greatly enhanced compared to normal subjects 12, 13, 58, 59 and its overexpression may be a major cause of lung damage in CF patients. We demonstrated here that a clinically relevant Bc strain (heat-inactivated) can activate NF-κB in airway epithelial cells, leading to increased IL-8 production. Stimulation of the cells with TNF-α, IL-1β or bacteria induced a higher level of IL-8 expression in IB3-1 cells (Cftr−/−) than in C38 cells (Cftr-corrected), in agreement with previous reports using these cell lines or primary cells 60, 61. Although Cftr knockout mice can be used as a model for Bc infection in CF 62, 63, 64, they are unfortunately not suitable for assessing our HD-Ad-IL-8RNAi. There is no bona fide mouse homologue of human IL-8, and our IL-8 target sequence does not share sequence homology with rodent functional IL-8 homologues, such as MIP-2, KC or CINC-1. A large animal model may be useful in preclinical analysis of the HD-Ad-IL-8RNAi.
Chi et al 65 demonstrated that shRNA-mediated gene silencing of mRNAs is highly gene-specific and does not produce secondary effects, as detectable by genome-wide expression profiling. However, it is possible that not all sequences are gene-specific, and therefore each shRNA should be experimentally examined for nonspecific effects. Although we cannot rule out all genome-wide effects from the experiments performed here, the HD-Ad-IL8RNAi did not appear to affect the NF-κB pathway, which regulates transcription of many cytokine genes including IL-8, nor did it affect IL-6 production, which is also regulated by the NF-κB pathway. Use of a control HD-Ad (expressing lacZ) revealed that neither IL-8 nor IL-6 were induced by the Ad particle itself in cultured cells, although this does not rule out the possibility of inducing inflammatory cytokines in other cell types, such as macrophages 66.
Since HD-Ads can effectively transduce cells in vivo and in vitro, a vector targeting the IL-8 mRNA could have important clinical implications. IL-8 is an essential component of the host defense system and it may not be a viable approach to block its activity systemically. Local administration of HD-Ad-IL8RNAi, for example to the lung, may be more practical. In addition, many delivery methods for gene therapy exacerbate inflammation. Our vector could be combined with other gene therapy vectors to minimize host inflammatory responses.
Schematic diagram of the viral vector construction. A 600 bp fragment with mouse U6 promoter and IL-8 siRNA expression fragment was inserted into viral vector pC4HSU at the AscI site. Then viral vector was digested by PmeI to expose the viral ITRs, and used to transfect 293cre4 cells.
References
Olson TS, Singbartl K, Ley K . L-selectin is required for fMLP- but not C5a-induced margination of neutrophils in pulmonary circulation. Am J Physiol 2002; 282:R1245–52.
Bonfield TL, Panuska JR, Konstan MW, et al. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med 1995; 152:2111–8.
Mahida YR, Ceska M, Effenberger F, et al. Enhanced synthesis of neutrophil-activating peptide-1/interleukin-8 in active ulcerative colitis. Clin Sci (Lond) 1992; 82:273–5.
Loetscher P, Dewald B, Baggiolini M, Seitz M . Monocyte chemoattractant protein 1 and interleukin 8 production by rheumatoid synoviocytes. Effects of anti-rheumatic drugs. Cytokine 1994; 6:162–70.
Kukielka GL, Smith CW, LaRosa GJ, et al. Interleukin-8 gene induction in the myocardium after ischemia and reperfusion in vivo. J Clin Invest 1995; 95:89–103.
Moss RB, Kurup VP, Knutsen AP, et al. Significant microbiological effect of inhaled tobramycin in young children with cystic fibrosis. Clin Infect Dis 2003; 37:S225–64.
Ordonez CL, Henig NR, Mayer-Hamblett N, et al. Inflammatory and microbiologic markers in induced sputum after intravenous antibiotics in cystic fibrosis. Am J Respir Crit Care Med 2003; 168:1471–5.
George J . Gene therapy progress and prospects: Adenoviral vectors. Gene Ther 2003; 10:1135–41.
Strieter RM . Interleukin-8: a very important chemokine of the human airway epithelium. Am J Physiol 2002; 283:L688–9.
Li J, Kartha S, Iasvovskaia S, et al. Regulation of human airway epithelial cell IL-8 expression by MAP kinases. Am J Physiol 2002; 283:L690–9.
Reddi K, Phagoo SB, Anderson KD, Warburton D . Burkholderia cepacia-induced IL-8 gene expression in an alveolar epithelial cells Line: signaling through CD14 and mitogen-activated protein kinase. Pediatr Res 2003; 54:297–305.
Eidelman O . Control of the Proinflammatory State in Cystic Fibrosis Lung Epithelial cell by Genes from the TNF-αR/NF-κB Pathway. Mol Med 2001; 7:523–34.
Li J, Johnson XD, Iazvovskaia S, et al. Signaling intermediates required for NF-kappa B activation and IL-8 expression in CF bronchial epithelial cells. Am J Physiol 2003; 284:L307–15.
Aderem A, Ulevitch RJ . Toll-like receptors in the induction of the innate immune response. Nature 2000; 406:782–7.
Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F . Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem 1999; 274:10689–92.
Zhang G, Ghosh S . Toll-like receptor-mediated NF-kappaB activation: a phylogenetically conserved paradigm in innate immunity. J Clin Invest 2001; 107:13–9.
Gewirtz AT . Intestinal epithelial toll-like receptors: to protect. And serve? Curr Pharm Des 2003; 9:1–5.
Sekido N, Mukaida N, Harada A, et al. Prevention of lung reperfusion injury in rabbits by a monoclonal antibody against interleukin-8. Nature 1993; 365:654–7.
Mian BM, Dinney CPN, Bermejo CE, et al. Fully human anti-interleukin 8 antibody inhibits tumor growth in orthotopic bladder cancer xenografts via down-regulation of matrix metalloproteases and nuclear factor-kappaB. Clin Cancer Res 2003; 9:3167–75.
Wada T, Tomosugi N, Naito T, et al. Prevention of proteinuria by the administration of anti-interleukin 8 antibody in experimental acute immune complex-induced glomerulonephritis. J Exp Med 1994; 180:1135–40.
Auten RL, Richardson RM, White JR, et al. Nonpeptide CXCR2 antagonist prevents neutrophil accumulation in hyperoxia-exposed newborn rats. J Pharmacol Exp Ther 2001; 299:90–5.
Sharp P, Zamore PD . Molecular biology. RNA interference. Science 2000; 287:2431–3.
Elbashir SM, Harborth J, Lendeckel W, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001; 411:494–8.
Elbashir SM, Harborth J, Weber K, Tuschl T . Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods 2002; 26:199–213.
Sui G, Soohoo C, Affar el B, et al. A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc Natl Acad Sci U S A 2002; 99:5515–20.
Yu JY, DeRuiter SL, Turner DL . RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci U S A 2002; 99:6047–52.
Fire A, Xu S, Montgomery MK, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998; 391:806–11.
Jacque JM, Triques K, Stevenson M . Modulation of HIV-1 replication by RNA interference. Nature 2002; 418:435–8.
Oshiumi H, Begum NA, Masumoto M, Seya T . RNA interference for mammalian cells. Nippon Yakurigaku Zasshi 2002; 120:91–5.
Parks R . Improvements of Adenoviral Vector Technology: Overcoming Barriers for Gene Therapy. Clin Genet 2000; 58:1–11.
Flotte TR, Afione SA, Solow R, et al. Expression of the cystic fibrosis transmembrane conductance regulator from a novel adeno-associated virus promoter. J Biol Chem 1993; 268:3781–90.
Zeitlin PL, Lu L, Rhim J, et al. A cystic fibrosis bronchial epithelial cell line: immortalization by adeno-12-SV40 infection. Am J Respir Cell Mol Biol 1991; 4:313–9.
Ng P, Parks RJ, Graham FL . Preparation of helper-dependent adenoviral vectors. Meth Mol Med 2002; 69:371–88.
Kramer A, Keller W . Preparation and fractionation of mammalian extracts active in pre-mRNA splicing. Meth Enzymol 1990; 181:3–19.
Black HR, Yankaskas JR, Johnson LG, Noah TL . Interleukin-8 production by cystic fibrosis nasal epithelial cells after tumor necrosis factor-alpha and respiratory syncytial virus stimulation. Am J Respir Cell Mol Biol 1998; 19:210–5.
Richmond A . Nf-kappa B, chemokine gene transcription and tumour growth. Nat Rev Immunol 2002; 2:664–74.
Baggiolini M . Chemokines in pathology and medicine. J Intern Med 2001; 250:91–104.
Luster AD . Chemokines—chemotactic cytokines that mediate inflammation. N Engl J Med 1998; 338:436–45.
Mukaida N . Pathophysiological roles of interleukin-8/CXCL8 in pumonary disease. Am J Physiol 2003; 284:L566–77.
Payne AS, Cornelius LA . The role of chemokines in melanoma tumor growth and metastasis. J Invest Dermatol 2002; 118:915–22.
Tullus K, Noack GW, Burman LG, et al. Elevated cytokine levels in tracheobronchial aspirate fluids from ventilator treated neonates with bronchopulmonary dysplasia. Eur J Pediatr 1996; 155:112–6.
Boekholdt SM, Peters RJ, Hack CE, et al. IL-8 plasma concentrations and the risk of future coronary artery disease in apparently healthy men and women: the EPIC-Norfolk prespective population study. Arterioscler Thromb Vasc Biol 2004; 24:1503–8.
Li A, Dubey S, Varney ML, Dave BJ, Singh RK . IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol 2003; 170:3369–76.
Simonini A, Moscucci M, Muller DW, et al. IL-8 is an angiogenic factor in human coronary atherectomy tissue. Circulation 2000; 101:1519–26.
Ostergaard C, Yieng-Kow RV, Larsen CG, et al. Treatment with a monoclonal antibody to IL-8 attenuates the pleocytosis in experimental pneumococcal meningitis in rabbits when given intravenously, but not intracisternally. Clin Exp Immunol 2000; 122:207–11.
Yang XD, Corvalan JR, Wang P, Roy CM, Davis CG . Fully human anti-interleukin-8 monoclonal antibodies: potential therapeutics for the treatment of inflammatory disease states. J Leukoc Biol 1999; 66:401–10.
Huang S, Mills L, Mian B, et al. Fully humanized neutralizing antibodies to interleukin-8 (ABX-IL8) inhibit angiogenesis, tumor growth, and metastasis of human melanoma. Am J Pathol 2002; 161:125–34.
Koehler DR, Hitt MM, Hu J . Challenges and strategies for cystic fibrosis lung gene therapy. Mol Ther 2001; 4:84–91.
Arts GJ, Langemeijer E, Tissingh R, et al. Adenoviral vectors expressing siRNAs for discovery and validation of gene function. Genome Res 2003; 13:2325–32.
Shen C, Buck AK, Liu X, Winkler M, Reske SN . Gene silencing by adenovirus-delivered siRNA. FEBS Lett 2003; 539:111–4.
Tiscornia G, Singer O, Ikawa M, Verma IM . A general method for gene knockdown in mice by using lentiviral vectors expressing small interfering RNA. Proc Natl Acad Sci U S A 2003; 100:1844–8.
Xia H, Mao Q, Paulson HL, Davidson BL . siRNA-mediated gene silencing in vitro and in vivo. Nat Biotechnol 2002; 20:1006–10.
Hillgenberg M, Tonnies H, Strauss M . Chromosomal integration pattern of a helper-dependent minimal adenovirus vector with a selectable marker inserted into a 27.4-kilobase genomic stuffer. J Virol 2001; 75:9896–908.
Palmer D, Ng P . Improved system for helper-dependent adenoviral vector production. Mol Ther 2003; 8:846–52.
Kube D, Sontich U, Fletcher D, Davis PB . Proinflammatory cytokine responses to P. aeruginosa infection in human airway epithelial cell lines. Am J Physiol 2001; 280:L493–502.
Palfreyman RW, Watson ML, Eden C, Smith AW . Induction of biologically active interleukin-8 from lung epithelial cells by Burkholderia (Pseudomonas) cepacia products. Infect Immun 1997; 65:617–22.
Takeda K, Kaisho T, Akira S . Toll-like receptors. Annu Rev Immunol 2003; 21:335–76.
Armstrong DS, Grimwood K, Carlin JB, et al. Lower airway inflammation in infants and young children with cystic fibrosis. Am J Respir Crit Care Med 1997; 156:1197–204.
Muhlebach MS, Stewart PW, Leigh MW, Noah TL . Quantitation of inflammatory responses to bacteria in young cystic fibrosis and control patients. Am J Respir Crit Care Med 1999; 160:186–91.
Aldallal N, McNaughton EE, Manzel LJ, et al. Inflammatory response in airway epithelial cells isolated from patients with cystic fibrosis. Am J Respir Crit Care Med 2002; 166:1248–56.
Venkatakrishnan A, Stecenko AA, King G, et al. Exaggerated activation of nuclear factor-kappaB and altered IkappaB-beta processing in cystic fibrosis bronchial epithelial cells. Am J Respir Cell Mol Biol 2000; 23:396–403.
Davidson DJ, Dorin JR, McLachlan G, et al. Lung disease in the cystic fibrosis mouse exposed to bacterial pathogens. Nat Genet 1995; 9:351–7.
Koehler DR, Sajjan U, Chow Y-H, et al. Protection of Cftr knockout mice from acute lung infection by a helper-dependent adenoviral vector expressing Cftr in airway epithelia. Proc Natl Acad Sci U S A 2003; 100:15364–9.
Sajjan U, Thanassoulis G, Cherapanov V, et al. Enhanced susceptibility to pulmonary infection with Burkholderia cepacia in Cftr−/− mice. Infect Immun 2001; 69:5138–50.
Chi JT, Chang HY, Wang NN, et al. Genomewide view of gene silencing by small interfering RNAs. Proc Natl Acad Sci U S A 2003; 100:6343–6.
Higginbotham JN, Seth P, Blaese RM, Ramsey WJ . The release of inflammatory cytokines from human peripheral blood mononuclear cells in vitro following exposure to adenovirus variants and capsid. Hum Gene Ther 2002; 13:129–41.
Acknowledgements
We thank Karen Kwon for technical assistance and Yang Shi (Harvard Medical School, Boston, MA) for providing pBS/U6. This work was funded by operating grants from the Canadian Cystic Fibrosis Foundation (to Jim HU), an operating grant from the Foundation Fighting Blindness-Canada (To Jim HU) and operating grants from the Canadian Institutes of Health Research (to Jim HU and A. Keith TANAWELL). and Jim HU is a CCFF Scholar and holds a Premier's Research Excellence Award of Ontario Canada.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Rights and permissions
About this article
Cite this article
CAO, H., WANG, A., MARTIN, B. et al. Down-regulation of IL-8 expression in human airway epithelial cells through helper-dependent adenoviral-mediated RNA interference. Cell Res 15, 111–119 (2005). https://doi.org/10.1038/sj.cr.7290275
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7290275
Keywords
This article is cited by
-
Immunomodulation by cannabidiol in bovine primary ruminal epithelial cells
BMC Veterinary Research (2023)
-
Biosynthetic flexibility of Pseudomonas aeruginosa leads to hydroxylated 2-alkylquinolones with proinflammatory host response
Communications Chemistry (2023)
-
Effects of transgenic sterilization constructs and their repressor compounds on hatch, developmental rate and early survival of electroporated channel catfish embryos and fry
Transgenic Research (2015)
-
Knockdown of ZNF403 inhibits cell proliferation and induces G2/M arrest by modulating cell-cycle mediators
Molecular and Cellular Biochemistry (2012)
-
Gene therapy: light is finally in the tunnel
Protein & Cell (2011)

{kind=link}