
ABSTRACT
Pancreatic cancer is a challenging disease for patients, doctors and researchers who for decades have searched for a cure for this deadly malignancy. Although existing mouse models of pancreatic cancer have shed light on the mechanistic basis of the neoplastic conversion of the pancreas, their impact in terms of offering new diagnostics and therapeutic modalities remains limited. Chronic pancreatitis is an inflammatory disease of the pancreas that is associated with a gradual damage of the organ and an increased risk of developing neoplastic lesions. In this review, we propose that detailed studies of chronic inflammatory processes in the pancreas will provide insights into the evolution of pancreatic cancer. This information may prove useful in the design of effective therapeutic strategies to battle the disease.
Similar content being viewed by others


INTRODUCTION
Pancreatic ductal adenocarcinoma is the most common among pancreatic tumors and one of the most fatal human cancers. The mortality rate among patients with pancreatic cancer is greater than 90%, and all attempts to change these overwhelming statistics have failed. Pancreatic cancer aggressively metastasizes even when primary lesions are very minor, forcing the patients to succumb to the disease despite successful surgical removal of the primary tumor. Much is known about the genetics and pathogenesis of this disease due to extensive studies on pancreatic samples from patients with pancreatic cancer, laboratory work done on pancreatic cancer cell lines, and several mouse models of pancreatic cancer. All of this knowledge has yet to provide an effective cure for this disease. The reason for the lack of successful treatment may be due to the fact that although we have a fairly detailed understanding of the general path of pancreatic cancer progression, we still do not know how this cancer originates and how to detect its development early enough for the treatment to be effective. This knowledge could be obtained by monitoring patients with known predispositions to the development of pancreatic cancer. Though logical, the main challenge for this approach is the lack of known markers for early stages of disease. A mouse model of a disease predisposing to pancreatic cancer could help in tackling this challenge by offering experimental access to the early stages of pancreatic cancer development.
THE PANCREAS: ANATOMY, HISTOLOGY AND PATHOLOGY
The pancreas is an accessory organ of the digestive system, and plays an essential role in the process of food digestion and maintenance of homeostasis in the absorption and release of glucose. The first function is achieved by a complex system of the exocrine pancreas which consists of acinar and ductal cells, and the latter by the endocrine pancreas organized into the islets of Langerhans. Acinar cells produce digestive enzymes such as trypsin, elastase and amylase, and store them in the zymogen granules in the cytosol as inactive enzyme forms called zymogens. In response to specific signals such as the presence of food in the digestive system, acinar cells secrete zymogens into the ductal system which consists of numerous ducts and ductules lined by ductal epithelial cells. Pancreatic secretions are transported through the ductal system to the duodenum where lower pH and the presence of activating enzymes such as enterokinase initiate a proteolytic cascade by activating trypsin. The exocrine function of the pancreas provides the organism with the means to deal with food absorption and the control of this process is partially achieved by the endocrine component. The endocrine pancreas is composed of the islets of Langerhans which consist mainly of three different cell types: α-cells that produce glucagon, β-cells that produce insulin and γ-cells that produce somatostatin. They all play an important role in regulating glucose homeostatis in the organism. Increased levels of glucose are a signal to secrete insulin which in turn controls glucose uptake. Glucagon induces release of glucose, and somatostatin regulates sensitivity of cells to insulin and glucagon.
Normal pancreatic function is susceptible to several disorders that can originate both from exocrine and endocrine compartments.
Diabetes
The most common disease of the endocrine pancreas is diabetes which affects 6.3% of the population in the United States. Diabetes can be manifested as type I diabetes in which β-cells are destroyed by autoimmune response, or as more common type II diabetes in which patients develop insulin resistance despite initial normal insulin production. Both types of diabetes disrupt the insulin control over the glucose levels and adsorption, a condition which in critical situations may be fatal. Other diseases of the endocrine pancreas including endocrine malignancies are not too common, frequently benign and in several cases can be successfully removed surgically 1.
Pancreatitis
The most prevalent disorder of the exocrine pancreas is pancreatitis 2. Pancreatitis can be acute resulting from severe pancreatic insufficiency most frequently due to high alcohol consumption. The pathogenesis of acute pancreatitis involves edematous changes in the acinar cell compartment accompanied by acute inflammatory infiltration and a certain degree of necrosis of the acinar cells 2. The integrity of the organ is fully restored after an episode of acute pancreatitis and the next episodes can be prevented or diminished by dietary restrictions. Pancreatitis may, however, be manifested as a chronic disease causing irreversible damage to the pancreatic parenchyma. Although it remains debatable whether the chronic form originates from the acute form or represents a distinct physiological disorder, the clinical manifestations of both diseases are very similar 2. Chronic pancreatitis however cannot be cured, and it has been linked to the most fatal disease of the pancreas which is pancreatic ductal adenocarcinoma 3.
Gradual destruction of the exocrine pancreas in chronic pancreatitis is caused by extensive chronic inflammation that can be characterized by the infiltration of T lymphocytes and macrophages, and development of the fibrotic reaction 4. As in many human inflammatory diseases, the degradative process is thought to be instigated by the injury of epithelium and the resulting recruitment of the inflammatory cells 5. The causative factors of pancreatic injury may be a prolonged period of alcohol abuse (alcoholic chronic pancreatitis), genetic predisposition (hereditary pancreatitis or autoimmune pancreatitis), blocked or narrowed pancreatic duct (obstructive pancreatitis) or other unknown factors (idiopathic pancreatitis) 6, 7. Other causes of chronic pancreatitis include pathological conditions such as cystic fibrosis, hypercalcemia, hyperlipidemia or hypertriglyceridemia 4. The clinical manifestations of chronic pancreatitis involve strong abdominal pain, nausea, vomiting, loss of weight and often secondary diabetes as a result of the inflammatory destruction of endocrine pancreas 8. At the histological level, chronic pancreatitis is manifested by the gradual loss of exocrine and endocrine cells and the development of fibrosis. Fibrosis can be defined as the formation of extracellular matrix in the interstitial spaces and in areas where the exocrine compartment, mainly acinar cells, disappear. Progressive fibrosis gradually leads to the loss of lobular architecture of the pancreas and damage of ductal and endocrine system resulting in pancreatic insufficiency 6.
In conclusion, chronic pancreatitis, like other chronic diseases, causes irreversible injury of the organ and its replacement by scar tissue as a result of the fibrotic reaction. Patients with chronic pancreatitis have an increased risk of developing pancreatic cancer with a rate 15- to 16-fold greater than that of the general population 9. Interestingly, pancreatic cancer may also lead to secondary chronic pancreatitis and secondary diabetes, which is linked to a very poor prognosis for these patients.
Pancreatic cancer
Pancreatic cancer is a serious health problem due not to its prevalence, but to its outcome. Of ∼30,000 new reported cases in the USA each year, less than 1% of patients will survive 5 years 10. The insidious nature of pancreatic cancer stems largely from the inability to clinically detect its earliest forms. Late detection is caused by the paucity of information regarding risk factors that may predispose patients to pancreatic cancer. Environmental factors such as smoking and alcohol consumption have been implicated as predisposing factors to pancreatic cancer 11. Chances to develop this malignancy increase highly with age, reaching a 40-fold increased risk by the age of 80 12. Genetically, certain germ line mutations in tumor suppressor genes such as BRCA1, LKB1, MLH1 and CDKN2A have been shown to increase the risk of pancreatic cancer development. However, the penetrance of these genetic lesions is comparable to that observed in sporadic cases of pancreatic cancer 12, 13. Therefore, these environmental and genetic criteria do not provide a solid basis for accurate risk assessment.
Significant contributions to the field of pancreatic cancer genetics have been made by the analysis on resected pancreatic tissue from cancer patients. These studies have allowed for the compilation of a temporal map of the genetic lesions within the ductal epithelium occurring during the course of tumor progression. Based on these studies, it has been shown that pancreatic adenocarcinoma progresses through a series of advancing morphological stages, known as pancreatic intraepithelial neoplasias (PanINs) 14. First, low cuboidal ductal cells become tall columnar due to extensive mucin production. Next, these epithelial cells start to show a certain degree of nuclear atypia and an enhanced proliferation rate which progressively increases and eventually leads to ductal cell shedding into the lumen of the ducts which facilitates tumor metastasis. These morphological alterations correlate with increasing genetic abnormalities. It has been shown that within the PanIN classification system, activating KRas mutations are the earliest and most conserved mutations identified 15. The activation of KRas is followed by the sequential loss of the tumor suppressors, p16, p53, and DPC4 and upregulation of telomerase 15. Early overexpression of epidermal growth factor receptor (HER-2/Neu) and the cell cycle regulator p21 (WAF1/CIP1) has also been reported 16, 17. However, while these studies provide a retrospective look at genetic lesions that are of importance in pancreatic cancer, they still rely on the resection of pancreatic tissue that has been affected by clinically evident disease.
The concept of pancreatic adenocarcinoma originating from the ductal epithelial cells has recently been questioned by findings derived from the mouse models of pancreatic cancer. Transgenic expression of oncogenes from developmentally active promoters gave rise to pancreatic cancer in the mouse suggesting the role of the pancreatic progenitor cells as an origin of cancer 18, 19, 20. Additionally, expression of oncogenes in acinar cells led to the development of pancreatic tumors with the ductal phenotype (see below) 21. Moreover, focal expression of non-ductal cell markers, such as endocrine and acinar enzymes, as well as the reemergence of expression of developmental signaling pathways has been detected in the samples of human ductal adenocarcinoma 12. Altogether these data illustrate the gaps in our knowledge of the origin of pancreatic cancer and emphasize the need for a model system that could address the issues of pancreatic cancer initiation and development. Ideally, such a system could provide markers for the inciting events of cancer development and allow for the early detection of the disease.
MOUSE MODELS OF PANCREATIC CANCER
Creation of a mouse model that will recapitulate PanIN lesions observed in humans has been a long awaited experimental goal. Despite well-established knowledge about the progressive genetic lesions in human cancer, many attempts to use this information and recapitulate pancreatic cancer development in the mouse led to the unsatisfactory results 22. An early model developed by Sandgren et al. was generated by the overexpression of tumor growth factor alpha (TGFα) in the acinar cells, and led to metaplastic transition of the acinar compartment into the pseudo-ductular structures. This was accompanied by the development of extensive fibrosis in the pancreatic parenchyma 19. Although this model failed to induce PanIN lesions, it showed that the mouse pancreas is sensitive to extensive TGFα signaling. This sensitivity might be analogous to the early stages of human pancreatic cancer development in which overexpression of the receptor for this ligand, namely EGF receptor, is detected. Interestingly, the introduction of the second genetic lesion observed in human cancer, p53 mutation, led to the progression of the phenotype towards invasive cancer which expressed ductal cell markers. However this transition did not occur through PanINs 21. Subsequent trials to recapitulate the progression of a human pancreatic ductal adenocarcinoma in the mouse focused on the introduction of the most frequent genetic abnormality in pancreatic cancer, a mutated, constitutively active KRas. This has been achieved by overexpression of activated KRas specifically in the acinar or in the ductal cells by using tissue-specific promoters 18, 20. These two models also failed to recapitulate PanIN formation, resulting in mild inflammatory phenotype when activated KRas was expressed in the ductal cells, or hyperplastic and dysplastic phenotype of acinar cells when activated KRas was expressed in the acinar cells. The lack of phenotype in mice harboring ductal-specific expression of activated KRas is rather unexpected given that the ductal cell has been considered as an origin of pancreatic ductal adenocarcinoma.
Recently, Tuveson and colleagues have created a model in which the expression of activated KRas is directed to the progenitor cells of the mouse pancreas 23. This has been achieved by the knock-in of an activating mutation in endogenous KRas through Cre-mediated recombination in cells expressing Pdx1, a transcription factor that is expressed during pancreatic development 23. This approach has proven to be successful and resulted in the full recapitulation of the PanIN lesions and their progression towards invasive carcinoma with characteristics mimicking human disease. It remains to be determined whether the success of this approach can be attributed to the embryonic expression of activated KRas or to the endogenous levels of expression of the mutant KRas, or both. Certainly this model provides an invaluable tool to test new therapeutics. However, its adequacy in terms of analysis of the initiating events of pancreatic cancer is less clear.
CHRONIC INFLAMMATION AND CANCER
The link between chronic inflammation and tumorigenesis has been recognized in many pathological conditions including esophageal cancer after Barrett's metaplasia, colorectal cancer after inflammatory bowel disease, ulcerative colitis and Crohn's disease or bladder cancer after schistosomasis and pancreatic ductal adenocarcinoma after chronic pancreatitis 5. Extensive inflammation during chronic inflammatory diseases exposes the organ tissue to the cytotoxic agents such as proinflammatory cytokines and reactive oxygen species. Their presence may lead to activation of cellular protective mechanisms, such as cell death and increased proliferation aiming at organ regeneration. Increased cell turnover in an environment rich in oxidative species may favor accumulation of DNA damage increasing chances of positive selection for mutations that confer growth advantage. The mechanisms by which inflammation affects tissue homeostasis are quite well established. However, the nature of the transition between chronic inflammation and cancer is still a subject of debate.
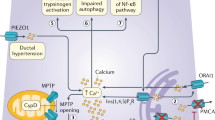
The pathogenesis of chronic pancreatitis is thought to follow a scenario described for other human chronic inflammatory conditions 9, 24, 25, 26. In this model, epithelial cells in response to the damaging event, such as infection or necrosis which in the pancreas can be caused by extensive tissue autodigestion by digestive enzymes released from the zymogen granules, express the transcription factor NFκB. Activation of NFκB has been shown to be an early response to inflammation in experimental models of pancreatitis and leads to the secretion of pro-inflammatory cytokines, such as TNFα and IL-1β 27, 28, 29. Concomitant activation of endothelial cells and recruitment of activated macrophages to the site of damage will enhance cytokine release and induce expression of acute phase genes such as COX-2 and IL-8 30, 31. Expression of acute phase genes together with the enhanced expression of adhesion molecules such as VCAM, ICAM, E- and P-selectins on endothelial cells serve as a signal for the recruitment of the leukocytes to the site of the damage 32. Moreover, enhanced production of prostaglandins and leukotriens mediated by COX-2 will result in increased vasopermeability leading to a more robust recruitment of the leukocytes. Leukocytes are the main source of reactive oxygen species and nitric oxide which, among many other functions, have a significant deleterious effect on DNA and serve as a strong inducer of apoptosis. Oxidative stress can also activate transcription factors such as AP1 and NFκB, which will lead to the activation of survival and growth promoting signaling pathways and further cytokine release 33. Increased mitogenic signals in an environment rich in oxidative species may create a selective pressure to acquire mutations favoring survival and uncontrolled proliferation which may give rise to cancer.
Fibrosis is a hallmark of human pancreatic adenocarcinoma and chronic pancreatitis. The source of fibrosis in the pancreas is fibroblast-like cells located peri-acinarily, called pancreatic stellate cells (PSCs). Under normal conditions these cells remain quiescent. However during inflammation, activating signals from cytokines, such as TNFα, and growth factors, such as TGF-β1, TGFα and PDGF, stimulate these cells to proliferate and differentiate into myofibroblasts 4. Myofibroblasts express α-smooth muscle actin and secrete extracellular matrix (ECM) proteins such as collagen types I and III, and fibronectin which replace the necrotic tissue 34. Initially this reaction is thought to help tissue regeneration. However, in the context of chronic inflammation, fibrosis takes over the normal tissue 35. Moreover, fibrosis is believed to additionally contribute to tissue damage since activated myofibroblasts can also secrete matrix metalloproteinases to destroy the normal ECM and deposit a newly formed fibrotic ECM components 6, 36. Activated fibroblasts can also be a source of growth factors which can support proliferation of the injured tissue facilitating neoplastic conversion 34.
Despite the above mentioned deleterious effect of chronic inflammation on tissue integrity, survey of the statistics concerning the association of inflammatory conditions with cancer shows that the percentage of patients with chronic inflammatory disease progressing to cancer is not that high. It is the highest for bronchitis and lung cancer – 11-24 % patients with bronchitis will develop lung cancer. For other types of chronic inflammatory diseases, including pancreatitis, statistics show that below 10% of patients will progress to cancer 9. These data emphasize the collaborative effect of genetic background and other environmental factors, such as tobacco smoking or viral infections, that may cause some patients to progress to cancer.
MOUSE MODEL OF HEREDITARY PANCREATITIS–WILL IT IMPROVE OUR UNDERSTANDING OF PANCREATIC CANCER?
In order to achieve an experimental system in which the early transitional stages from normal pancreatic parenchyma to a malignant phenotype can be recapitulated, we have decided to create an animal model that mimics a human disease highly predisposing patients to pancreatic carcinoma, namely hereditary pancreatitis. As it has already been mentioned, chronic inflammation of the pancreas has been shown through multiple statistical analyses to be associated with an increased susceptibility to pancreatic adenocarcinoma. However, the numerous, often unknown causes of sporadic chronic pancreatitis, and the confounding factors of alcohol consumption and smoking, have made the creation of an animal model that mimics this human condition a difficult task. We have decided to take advantage of the known genetics of the hereditary form of chronic pancreatitis, which results from a germline mutation in PRSS1, the gene that encodes human cationic trypsinogen 37. This mutation changes arginine 117 to histidine (R177H) removing a tryptic cleavage site on trypsin, and therefore eliminates a negative feedback loop by which active trypsin inactivates itself. Additionally, this mutation leads, in the case of human trypsinogen, to an increased activation of the enzyme by auto-cleavage of the inhibitory peptide 38. At the level of the pancreatic organ, the presence of mutant trypsin is believed to create a prolonged exposure of the pancreatic parenchyma to trypsin activity which results in tissue destruction and an inflammatory infiltration.
Hereditary pancreatitis is an autosomal dominant disorder with an ∼80% penetrance 37. It is a relatively rare disease with a very early onset, usually in childhood, and is manifested as repeating episodes of acute pancreatitis. Many patients with hereditary pancreatitis progress to severe chronic pancreatitis and often to pancreatic cancer. The risk of pancreatic cancer for these patients is ∼53-fold higher than for healthy patients and the lifetime incidence rate is increased to 40% 39. These high statistics are believed to result from the early onset of the disease and the subsequent long-term exposure of the pancreas to inflammation.
Our model has incorporated the causative lesion, R122H mutation in the mPRSS1 gene which encodes mouse cationic trypsinogen. R122H mutation in the mouse trypsinogen corresponds to R117H mutation in the human homologue. The mutant mPRSS1 has been expressed in the mouse from the elastase promoter which drives the expression of the transgene specifically in the acinar cells. The initial characterization of transgenic animals revealed an inflammatory response of the pancreas with a 75% penetrance, closely mimicking that of the human disease. A subset of animals with this advanced disease displayed regions of acinar to ductal transdifferentiation, suggestive of a precursor to neoplasia. Currently we are in the process of analyzing the signaling and mechanistic pathways that are altered in pancreatic cell compartments in response to inflammation. Our preliminary results suggest that acinar and ductal cell compartments show dramatically different responses to chronic inflammatory stimuli indicating that this model encompasses a complex global response of the pancreas to inflammation. Further environmental and genetic manipulations of this model, are currently underway to enhance the progression of the phenotype and our understanding of the role of chronic inflammation in neoplastic conversion within the pancreas.
FUTURE PERSPECTIVES
Pancreatic cancer is a deadly disease known for its rapid progression and the lack of effective treatments. The generation of a hereditary pancreatitis model provides a platform for the elucidation of the inciting events in the development of pancreatic carcinoma. In this model the uncontrolled activation of a mutant trypsin results in a chronic inflammatory response that progresses to metaplastic lesions in the pancreatic cell compartments. We plan to explore the effects of epigenetic and genetic changes on the rate and extent of disease progression in this model. Furthermore, the establishment of a serum proteomic profile corresponding to different disease stages in our mouse model may contribute to the development of diagnostic tools for the detection of pancreatic cancer as well as for risk prediction. Finally, the pharmacological intervention with disease progression in our model should provide a framework for the preclinical identification of effective therapeutic targets.
References
Akerstrom G, Hellman P, Hessman O, Osmak L . Surgical treatment of endocrine pancreatic tumours. Neuroendocrinology 2004; 80:62–6.
Talamini G, Falconi M, Bassi C, et al. Chronic pancreatitis: relationship to acute pancreatitis and pancreatic cancer. Jop 2000, 1:69–76.
Lowenfels AB, Maisonneuve P, Cavallini G, et al. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med 1993, 328:1433–7.
Ebert M, Schandl L, Schmid RM . Differentiation of chronic pancreatitis from pancreatic cancer: recent advances in molecular diagnosis. Dig Dis 2001, 19:32–6.
Orlando RC . Mechanisms of epithelial injury and inflammation in gastrointestinal diseases. Rev Gastroenterol Disord 2002, 2:S2–8.
Kloppel G, Detlefsen S, Feyerabend B . Fibrosis of the pancreas: the initial tissue damage and the resulting pattern. Virchows Arch 2004, 445:1–8.
Stevens T, Conwell DL, Zuccaro G . Pathogenesis of chronic pancreatitis: an evidence-based review of past theories and recent developments. Am J Gastroenterol 2004, 99:2256–70.
Etemad B, Whitcomb DC . Chronic pancreatitis: diagnosis, classification, and new genetic developments. Gastroenterology 2001, 120:682–707.
Vakkila J, Lotze MT . Inflammation and necrosis promote tumour growth. Nat Rev Immunol 2004, 4:641–8.
Warshaw AL, Fernandez-del Castillo C . Pancreatic carcinoma. N Engl J Med 1992, 326:455–65.
Talamini G, Bassi C, Falconi M, et al. Alcohol and smoking as risk factors in chronic pancreatitis and pancreatic cancer. Dig Dis Sci 1999, 44:1303–11.
Bardeesy N, DePinho RA . Pancreatic cancer biology and genetics. Nat Rev Cancer 2002, 2:897–909.
Bardeesy N, Sharpless NE, DePinho RA, Merlino G . The genetics of pancreatic adenocarcinoma: a roadmap for a mouse model. Semin Cancer Biol 2001, 11:201–18.
Hruban RH, Adsay NV, Albores-Saavedra J, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol 2001, 25:579–86.
Hruban RH, Goggins M, Parsons J, Kern SE . Progression model for pancreatic cancer. Clin Cancer Res 2000, 6:2969–72.
Biankin AV, Kench JG, Morey AL, et al. Overexpression of p21 (WAF1/CIP1) is an early event in the development of pancreatic intraepithelial neoplasia. Cancer Res 2001, 61:8830–7.
Korc M, Chandrasekar B, Yamanaka Y, et al. Overexpression of the epidermal growth factor receptor in human pancreatic cancer is associated with concomitant increases in the levels of epidermal growth factor and transforming growth factor alpha. J Clin Invest 1992, 90:1352–60.
Brembeck FH, Schreiber FS, Deramaudt TB, et al. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res 2003, 63:2005–9.
Sandgren EP, Luetteke NC, Palmiter RD, Brinster RL, Lee DC . Overexpression of TGF alpha in transgenic mice: induction of epithelial hyperplasia, pancreatic metaplasia, and carcinoma of the breast. Cell 1990, 61:1121–35.
Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP . Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res 2003, 63:2016–9.
Wagner M, Greten FR, Weber CK, et al. A murine tumor progression model for pancreatic cancer recapitulating the genetic alterations of the human disease. Genes Dev 2001, 15:286–93.
Leach SD . Mouse models of pancreatic cancer: the fur is finally flying! Cancer Cell 2004, 5:7–11.
Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4:437–50.
Coussens LM, Werb Z . Inflammation and cancer. Nature 2002, 420:860–7.
Balkwill F, Mantovani A . Inflammation and cancer: back to Virchow? Lancet 2001, 357:539–45.
Clevers H . At the crossroads of inflammation and cancer. Cell 2004, 118:671–4.
Karin M, Cao Y, Greten FR, Li ZW . NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer 2002, 2:301–10.
Algul H, Tando Y, Schneider G, et al. Acute experimental pancreatitis and NF-kappaB/Rel activation. Pancreatology 2002, 2:503–9.
Steinle AU, Weidenbach H, Wagner M, Adler G, Schmid RM . NF-kappaB/Rel activation in cerulein pancreatitis. Gastroenterology 1999, 116:420–30.
Maier JA, Hla T, Maciag T . Cyclooxygenase is an immediate-early gene induced by interleukin-1 in human endothelial cells. J Biol Chem 1990, 265:10805–8.
Rodgers HC, Pang L, Holland E, et al. Bradykinin increases IL-8 generation in airway epithelial cells via COX-2-derived prostanoids. Am J Physiol Lung Cell Mol Physiol 2002, 283:L612–8.
Ardies CM . Inflammation as cause for scar cancers of the lung. Integr Cancer Ther 2003, 2:238–46.
Farrow B, Evers BM . Inflammation and the development of pancreatic cancer. Surg Oncol 2002, 10:153–69.
Jaster R . Molecular regulation of pancreatic stellate cell function. Mol Cancer 2004, 3:26.
Zimmermann A, Gloor B, Kappeler A, et al. Pancreatic stellate cells contribute to regeneration early after acute necrotising pancreatitis in humans. Gut 2002, 51:574–8.
Yokota T, Denham W, Murayama K, et al. Pancreatic stellate cell activation and MMP production in experimental pancreatic fibrosis. J Surg Res 2002, 104:106–11.
Whitcomb DC, Gorry MC, Preston RA, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 1996, 14:141–5.
Sahin-Toth M, Toth M . Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cationic trypsinogen. Biochem Biophys Res Commun 2000, 278:286–9.
Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst 1997, 89:442–6.
Acknowledgements
We thank members of the laboratory for helpful discussions and The Lustgarten Foundation for funding.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
JURA, N., ARCHER, H. & BAR-SAGI, D. Chronic pancreatitis, pancreatic adenocarcinoma and the black box in-between. Cell Res 15, 72–77 (2005). https://doi.org/10.1038/sj.cr.7290269
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7290269
Keywords
This article is cited by
-
Role of Hippo pathway dysregulation from gastrointestinal premalignant lesions to cancer
Journal of Translational Medicine (2024)
-
CD14/TLR4 priming potentially recalibrates and exerts anti-tumor efficacy in tumor associated macrophages in a mouse model of pancreatic carcinoma
Scientific Reports (2016)
-
Orthotopic inflammation-related pancreatic carcinogenesis in a wild-type mouse induced by combined application of caerulein and dimethylbenzanthracene
Tumor Biology (2015)
-
The proline TP53 variant stimulates likely lymphangiogenesis in an orthotopic mouse model of pancreatic cancer
British Journal of Cancer (2012)
-
Glucagon-like peptide-1 therapy and the exocrine pancreas: innocent bystander or friendly fire?
Diabetologia (2010)
