
Abstract
We report the first genetic analysis of free-ranging Asian elephants (Elephas maximus). We sampled 118 elephants from Sri Lanka, Bhutan/North India, and Laos/Vietnam by extracting DNA from dung, PCR amplifying and sequencing 630 nucleotides of mitochondrial DNA, including part of the variable left domain of the control region. Comparison with African elephant (Loxodonta africana) sequences indicated a relatively slow molecular clock in the Proboscidea with a sequence divergence of ≈1%/Myr. Genetic diversity within Asian elephants was low, suggesting a small long-term effective population size. Seventeen haplotypes were identified within Asian elephants, which clustered into two well-differentiated assemblages with an estimated Pliocene divergence of 2.5–3.5 million years ago. The two assemblages showed incomplete geographical partitioning, suggesting allopatric divergence and secondary admixture. On the mainland, little genetic differentiation was observed between elephant populations of Bhutan and India or Laos and Vietnam. A significant difference in haplotype frequencies but relatively weak subdivision was observed between the regions Bhutan–India and Laos–Vietnam. Significant genetic differentiation was observed between the mainland and Sri Lanka, and between northern, mid-latitude and southern regions in Sri Lanka.
Similar content being viewed by others
Introduction
The Asian elephant (Elephas maximus) once ranged from the Euphrates–Tigris rivers in the west to the Yangtze-Kiang river in the east (Olivier, 1978). Having been extirpated from approximately 85% of its historical range, it is presently classified as ‘threatened’ (Sukumar, 1989), and exists in a number of fragmented and isolated populations in South and South-east Asia (Fig. 1). The current population of Asian elephants is estimated at 35 000–50 000 individuals (Olivier, 1978; Sukumar, 1989).

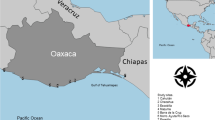
Sampling sites and elephant distribution. The number of samples successfully amplified from each location/geographical unit is indicated within parentheses. Inset: map of Asia with past (stippled) and present (shaded) distribution of elephants (from Santiapillai & Jackson, 1990). Map of Sri Lanka: shaded area represents the present distribution of elephants (from Santiapillai & Jackson, 1990). The dotted line demarcates the wet zone/dry zone boundary.
As in other parts of Asia, elephant populations in Sri Lanka have drastically declined in numbers and range in the past few decades. Elephants inhabited most of the island at the turn of the century, but were practically eliminated from the wet zone by hunting and changing land-use patterns during the colonial era (Jayewardene, 1994). The current elephant population in Sri Lanka is estimated at approximately 3000 animals (Jayewardene, 1994), which occupy mostly the dry zone of the country (Fig. 1). Two remnants of the wet zone populations survive as isolated groups in the Adam’s Peak and Walankanda areas (Fig. 1), and are estimated at 10–15 and five animals, respectively (Jayewardene, 1994).
Three subspecies of Asian elephants are currently recognized: E. maximus maximus in Sri Lanka, E. maximus indicus on the mainland, and E. maximus sumatranus in Sumatra (Shoshani & Eisenberg, 1982). Elephants from the mid-latitude range in Sri Lanka were once classified as a distinct subspecies E. maximus vilaliya (Deraniyagala, 1955) but were subsequently synonymized under the forma typica (McKay, 1973).
Previous genetic studies of Asian elephants have been limited to captive animals. Protein electrophoretic studies by Nozawa & Shotake (1990) and Hartl et al. (1995) found low levels of heterozygosity within Asian elephants, in contrast to a study by Drysdale & Florkiewicz (1989), which found relatively high levels of heterozygosity in both Asian and African elephants.
The study by Nozawa & Shotake (1990) also compared mainland (South India, Thailand and Nepal) and Sri Lankan animals, and found a complete allelic substitution at a single locus, thus corroborating the putative subspecific distinction between the two populations. Hartl et al. (1995) found allozyme variation between animals from the two populations at different loci from that reported by Nozawa & Shotake (1990). In contrast, a more recent evaluation of cytochrome b sequences found no support for a major separation of the Sri Lankan and mainland populations (Hartl et al., 1996). Thus, previous genetic studies of Asian elephants have yielded apparently conflicting results.
In this study we chose to analyse the highly variable d-loop region of mitochondrial DNA (mtDNA), which is particularly suitable for the genetic analysis of populations and closely related taxa. In addition to being ideal for studying the phylogeography of organisms, in species with a female-based social structure such as elephants, it can also be applied to the study of social organization (Fernando, 1998).
Obtaining samples of tissue, body fluids or hair, from free-ranging elephants is logistically difficult. A genetic study of free-ranging African elephants (Loxodonta africana) reported the use of biopsy darts to obtain skin samples (Georgiadis et al., 1994). As the threatened status of Asian elephants precluded the use of invasive sampling in this study, we chose dung as the source of DNA.
In this study, we address the extent and distribution of genetic diversity and phylogeography of Asian elephants, through sampling mtDNA from three widely separated geographical regions: Sri Lanka, Bhutan–India and Laos–Vietnam (Fig. 1). On a regional scale, we assess population subdivision and determine the population structure of elephants on the mainland and Sri Lanka. We use genetic data together with geological evidence to suggest an integrated interpretation of the genetic architecture of Asian elephants and demonstrate the utility of dung-extracted DNA analysis for studying a large population sample of a threatened species.
Materials and methods
Samples and study areas
Dung samples were obtained from a total of 123 elephants, consisting of 84 free-ranging elephants in Sri Lanka, five free-ranging and eight captive elephants from Bhutan, six captive elephants which originated from India, 13 free-ranging and three captive elephants from Laos, and four free-ranging elephants from Vietnam (Fig. 1). Although samples from Sri Lanka were obtained from locations throughout the present distribution of elephants in the country (Fig. 1), the current armed conflict in the northern and eastern regions limited access to some locations, resulting in under-sampling of some areas.
To validate the reliability of DNA sequences derived from dung, we compared sequences obtained from both blood and dung of two captive individuals. To confirm that sequences obtained from field-collected dung samples were of elephant origin, we compared them with the sequences obtained from the captive individuals. Four blood-extracted DNA samples were obtained from African elephants in USA zoos for use as an outgroup.
To assess DNA degradation prior to collection, dung boli from two free-ranging elephants observed defecating were left in the field and serially sampled at 5 and 25 min, 10 h, 2, 4 and 8 days after deposition. All other samples were randomly collected at a single point in time from each location. Twelve of the sampled dung boli were estimated to be over two weeks old, whereas the rest were estimated as having been deposited less than 48 h prior to collection. Stored samples were analysed at intervals of up to four years after collection to detect DNA degradation in storage.
Dung samples of approximately 5 g were collected from single boli, by scraping off the surface crust. Samples were placed in 15 mL tubes with 95% ethanol, sealed with parafilm, and stored at ambient temperature.
DNA extraction, amplification and sequencing
Approximately 0.5 g of each sample was treated with SDS and Proteinase K and DNA extracted with phenol/chloroform/iso-amyl-alcohol. Extracts were purified using QIAGEN spin columns and the manufacturer’s protocol, and stored at −20°C. A 630 bp segment of mtDNA was amplified using primers MDL 3 [5′-CCCACAATTAATGGGCCCGGAGCG-3′; based on a mitochondrial sequence from an Asian elephant (C. Wemmer, pers. com.)] and MDL5 [5′-TTACATGAATTGGCAGCCAACCAG-3′; based on a cytochrome b sequence from an African elephant in Irwin et al. (1991)]. The first 109 bp of the amplified segment coded for the C terminal of cytochrome b, the next 135 bp coded for threonine and proline tRNAs, and the rest was noncoding mitochondrial control region. PCR amplification was performed in 25 μL reactions using 1 μL DNA extract, 2 μL 100 mg mL–1 BSA, 2.5 μL 10× PCR buffer, 2.5 μL 8 m M dNTP mix (Promega, Madison, WI), 0.5 μL 10 μM primers, 0.1 μL Taq DNA polymerase (Perkin Elmer Cetus, Emeryville, CA), and 15.9 μL water. Reactions, in a Perkin Elmer Cetus programmable DNA Thermocycler, were preceded by a 4-min denaturation step at 95°C followed by 40 cycles of 1 min each at: 63°C annealing; 72°C extension; and 94°C denaturation. Amplifications were electrophoresed on 1% agarose, stained with ethidium bromide, bands visualized under UV and punched out with a pipette tip, melted in 50 μL H2O, and 2 μL used as template for a 50-μL secondary amplification with the same conditions and reactant concentrations. Reamplified PCR products were electrophoresed on 2% low-melt agarose, stained with ethidium bromide, product bands excised under UV, and purified using QIAGEN spin columns and the manufacturer’s protocol.
To guard against contamination, DNA extractions were conducted in a separate room from amplifications, using different sets of equipment. PCR reactions were set up in an UV sterilized hood and negative controls were conducted with every PCR reaction. Negative controls for primary amplifications were punched out in-line with the bands for the amplified samples and used as negative controls for secondary amplifications.
Sequencing was carried out at the University of Oregon Sequencing Facility, in an Applied Biosystems Inc. ABI 377 automated DNA sequencer using Dye Terminator Cycle Sequencing. Initially, all amplified products were sequenced in the forward direction using primer MDL 5 and each new haplotype identified was sequenced in the reverse direction using primer MDL 3. Of the amplified segment, 600 bp corresponding to bases 15 145–15 753 of human mitochondrial DNA (Arnason et al., 1996), were used in the analysis.
Data analysis
Sequences were aligned and edited with SEQUENCHER version 3.0 (Gene Codes Corporation). Genetic diversity within populations was estimated using haplotype (ĥ) and nucleotide (π^) diversity estimates at three scales: (i) total sample; (ii) between regions (Bhutan–India and Laos–Vietnam; mainland and Sri Lanka); and (iii) within Sri Lanka. Haplotype diversity was estimated according to the method of Nei (1987). A chi-squared test of independence using the Monte Carlo randomization method in the program CHIRXC (Zaykin & Pudovkin, 1993) was used to test for significance of differences in haplotype frequencies, in a pairwise comparison of locations. An unrooted parsimony network of haplotypes was constructed by hand, showing the number of mutational steps between haplotypes. The program DNASP (Rozas & Rozas, 1997) was used to calculate the average number of nucleotide differences k, average number of nucleotide substitutions per site using the Jukes and Cantor correction Dxy(JC), nucleotide diversity, and estimates of population subdivision at the nucleotide level using NST. Estimates of NST were calculated at two scales: (i) between regions (Bhutan–India and Laos–Vietnam; mainland and Sri Lanka); and (ii) within Sri Lanka. Phylogenetic trees were constructed using pairwise uncorrected sequence divergence estimates between haplotypes with Neighbour Joining (NJ) and Maximum Parsimony (MP) procedures with the program PAUP* (Swofford, 1998). Trees were rooted using the African elephants as an outgroup. The robustness of the phylogenetic hypothesis was tested as percentage recurrence of clusters based on 1000 bootstrapped replications with PAUP*.
Results
Sequences were obtained from all dung samples analysed, except for five samples that did not provide amplification products. Three of the samples that failed to amplify were estimated to have been collected over two weeks after deposition and the other two samples a few days after deposition. Identical sequences were obtained from dung and blood of captive animals, confirming that mtDNA sequences obtained from dung were the same as that obtained from blood, and that they were not artifactual. Sequences derived from field-collected dung samples and control sequences from zoo animals aligned unambiguously, indicating that the sequences obtained from field-collected dung samples were of elephant origin. Identical sequences were obtained from all samples of the two serially sampled dung piles, up to and including the last samples of the series (8 days), suggesting that dung left in the field for up to eight days provided DNA of sufficient integrity for PCR analysis. Samples that had been stored at ambient temperature in ethanol for up to four years also provided sequences with unambiguous alignments, indicating no detectable degradation over long periods of time.
Genetic variation
Between Asian and African elephants
Alignment of the Asian and African elephant sequences resulted in 54 polymorphic sites consisting of 52 substitutions and two insertion/deletions (indels) (data not shown). Twenty substitutions and the two indels were fixed between the genera. The observed transition/transversion ratio was 11.75. Of the variable sites, five were autapomorphies and 49 were phylogenetically informative with an average number of nucleotide substitutions per site between the two genera Dxy (JC)=0.06556.
Within Asian elephants
A total of 27 sites were found to be polymorphic (Table 1), with a single autapomorphy (position 431) and 26 phylogenetically informative sites. The transition/transversion ratio was 5.2, with all transversions in the control region. More than two different nucleotides were detected (C, T and G) at only one position (position 417). The total nucleotide diversity was π^=0.017 and average number of nucleotide differences was k=10.367. Fifteen haplotypes were identified within Asian elephants, and the total haplotype diversity was ĥ=0.871. Haplotype frequency values ranged from 0.263 for the most frequent haplotype (haplotype E), to 0.008 for five haplotypes that had single occurrences (Table 2).
Phylogenetic pattern
Haplotypes segregated into two distinct assemblages (Fig. 2), assemblage α with 7 haplotypes, and assemblage β with 10 haplotypes, with eight fixed substitutions separating them (excluding positions where homoplasy was assumed). Haplotype diversity was estimated at ĥ=0.698 and 0.806, nucleotide diversity π^=0.00238 and 0.00549, and average number of nucleotide differences k=1.431 and 3.293, for assemblages α and β, respectively. The average number of nucleotide substitutions between the two assemblages was Dxy (JC)=0.03135. Phylogenetic trees generated using NJ and MP methods were in complete agreement with each other, with a high level of bootstrap support for the two assemblages within Asian elephants (assemblage α 99%, assemblage β 77%) (data not shown). Thus, a phylogenetic discontinuity was observed within Asian elephants, with an estimated sequence divergence of 3.14% between two observed assemblages, which was approximately half that between Asian and African elephants (6.56%).

Parsimony network of Asian elephant haplotypes. Letters within circles denote haplotypes. Circles without letters denote haplotypes assumed but not observed in the present study. Letters in shaded circles denote mainland haplotypes, in striped circles haplotypes shared between the mainland and Sri Lanka, and in clear circles haplotypes limited to Sri Lanka. Dashes between haplotypes represent mutational steps between haplotypes, and the number adjacent denotes the base position of the polymorphism. Asterisks mark instances of assumed homoplasy. Position 417 contained a transition and a transversion, which are denoted as 417a and 417b, respectively.
Population structure
Within the mainland
Both assemblages α and β were observed on the mainland (Fig. 2), with frequencies of 0.945 and 0.054, respectively. Nucleotide diversity was highest in Bhutan, and haplotype diversity highest in Vietnam (Table 3)). Although a bias caused by sampling only captive animals cannot be excluded, genetic diversity was surprisingly lacking in India. Similar haplotype frequencies and relatively low subdivision was observed between adjacent countries within the mainland, suggesting single population units (between India and Bhutan: χ2=4.2, P=0.169; NST=0.166; between Laos and Vietnam: χ2=6.429, P=0.187; NST=0.148). Although haplotype frequencies differed significantly (χ2=27.26, P < 0.001), relatively weak subdivision (NST=0.1314) was observed between the regions Bhutan–India and Vietnam–Laos.
Mainland and Sri Lanka
Haplotype E was the most frequent haplotype in the total sample. Haplotypes E and L were the only haplotypes shared between the mainland and Sri Lanka, and were the most frequent haplotypes in the assemblages α and β, respectively. Together haplotypes E and L accounted for over 43% of the sampled individuals. Haplotype E was scored in 12 and 19 individuals, and haplotype L in one and 19 individuals from the mainland and Sri Lanka, respectively.
Genetic diversity indices were higher in Sri Lanka than on the mainland (Table 3). Haplotype frequencies differed significantly between the two regions (χ2=82.14, P < 0.001) and strong subdivision (NST=0.509) was observed, demonstrating relatively high genetic differentiation of Asian elephant populations between the mainland and Sri Lanka.
Within Sri Lanka
Sri Lankan populations were characterized by 10 private haplotypes (Table 3). Both assemblages α and β were observed in Sri Lanka (Fig. 2), with frequencies of 0.347 and 0.655, respectively. All observed individuals of assemblage α haplotypes were from the southern region, whereas assemblage β haplo- types were more widespread (Table 2). Haplotypes E (from assemblage α) and L (from assemblage β) were the most frequent haplotypes in Sri Lanka, both being scored in 19 individuals.
The wet zone populations represented by single samples from Adam’s Peak and Walankanda (Fig. 1) were of haplotypes E and G. Because they were a subset of the southern haplotypes (Table 2), they were included in the southern group in the analysis.
Comparison of indices of genetic differentiation found no major separations within the northern (sampling locations 1, 2, 3 and 6), mid-latitude (locations 4, 5, 7, 8, 9 and 10), and southern (locations 11, 12, 13, 14 and 15) regions (Fig. 1) (data not shown). However, significant differences in haplotype frequencies and relatively high subdivision was observed between them (Table 4), suggesting that Sri Lankan populations could be genetically divided into northern, mid-latitude and southern groups. Haplotype diversity was highest in the mid-latitude region, and nucleotide diversity highest in the south (Table 3).
Thus, our data suggest higher mtDNA diversity in Sri Lanka and strong population subdivision at the level of mtDNA between the mainland and Sri Lanka and within Sri Lanka.
Discussion
In the present study, we calculated a sequence divergence of 6.56% between the genera Elephas and Loxodonta. Dating the divergence of Elephas and Loxodonta lineages as 5–7 million years ago on palaeontological evidence (Maglio, 1973), and assuming a constant rate of evolution, the estimated rate of change in control region sequence between the two lineages is ≈1%/Myr. This is lower than the estimate of ≈8 to 15%/Myr for other terrestrial mammals, but comparable to that estimated for baleen whales ≈1%/Myr and dugong ≈2%/Myr (Garcia-Rodriguez et al., 1998). The observed lower rate in the Proboscidea could be explained by the reduction in the rate of the molecular clock with increase in body mass, longer generation time, and lower metabolic rate (Martin et al., 1993). The findings of the present study are consistent with previous allozyme findings which demonstrated a lower genetic divergence between the two Proboscidean genera, than between genera in other groups of mammals (Drysdale & Florkiewicz, 1989).
Population structure
We analysed the population structure of Asian elephants by hierarchical analysis of the data. Comparisons between geographical units suggested that a two-level analysis was appropriate: between the mainland and Sri Lanka, and within Sri Lanka.
Mainland and Sri Lanka
Although two of the 15 haplotypes observed by us were common to the mainland and Sri Lanka (Fig. 2), we observed high genetic divergence between the two regions (Table 3), which is in accord with the results of previous allozyme studies (Nozawa & Shotake, 1990; Hartl et al., 1995). In contrast, the study of the cytochrome b sequence by Hartl et al. (1996) found low differentiation between the two populations. Such a finding is difficult to reconcile with the allozyme data and our findings, but may in part be explained by the expected lower variability of the cytochrome b region.
Our finding of two shared haplotypes between the mainland and Sri Lanka supports the view of Hartl et al. (1996) that a separate subspecies designation of mainland and Sri Lankan elephants was not justified. However, subspecies and distinct taxa may exhibit shared mtDNA polymorphisms (Dowling et al., 1992; Ramey, 1995), and the distinction between subspecies should be based on concordant distribution of multiple, independent and genetically based traits. Previous allozyme findings of high genetic differentiation (Nozawa & Shotake, 1990; Hartl et al., 1995), and observed morphological differences between the two populations (Deraniyagala, 1955), warrant further study using nuclear DNA markers and morphological data, before concluding that the subspecific designation between mainland and Sri Lankan elephants is not justified.
Within Sri Lanka
Relatively high levels of differentiation in mtDNA were observed between the northern, mid-latitude and southern regions of the island. As there are no extrinsic barriers to gene flow within Sri Lanka, the observed subdivision needs to be explained by an alternate mechanism. Female elephants are strongly philopatric to their groups and home ranges (Sukumar, 1989). Radio tracking studies in southern Sri Lanka have shown that females have small home ranges [range = 20 570 ha–4672 ha, SE=3392 ha (Fernando, 1998)], and genetic analysis of female social groups has confirmed that female social organization is matriarchal (Fernando, 1998). As the mode of inheritance of mtDNA is clonal and matrilineal, the observed social organization and behaviour of female groups could explain the subdivision observed in the present study.
Phylogeography
A phylogenetic discontinuity of two assemblages of mtDNA haplotypes with a sequence divergence of 3.1% corresponding to a divergence time of 2.5–3.5 Myr ago and incomplete geographical partitioning of the two assemblages was observed in the present study. The possible evolutionary circumstances that could give rise to such a phylogeographic pattern are: (i) introgression of mtDNA through hybridization with a closely related species; (ii) incomplete lineage sorting; and (iii) ancestral isolation in allopatry with secondary admixture.
A hypothesis of introgression from a related species requires the contemporaneous existence of a conspecific. According to palaeontological evidence presented by Maglio (1973), the Elephas lineage originated in Africa and diverged into a species complex with an Asiatic and an African–European branch. Progenitors of the Asian branch invaded Asia in the late Pliocene and gave rise to the present form E. maximus and a number of other now extinct species. In early Pleistocene a second emigration out of Africa of the Elephas lineage occurred through E. namadicus. This species rapidly expanded in range and became dominant, occurring as the only elephant in parts of Asia but went extinct in late Pleistocene (Maglio, 1973). Thus, the phylogenetic discontinuity observed within Asian elephants in the present study, could have resulted from introgression of DNA from E. namadicus to E. maximus. However, this hypothesis predicts a greater than observed divergence between the two assemblages, as the divergence of the Asian and African branches of Elephas occurred soon after the divergence of the Elephas and Loxodonta lineages (Maglio, 1973). An alternative hypothesis is that the observed phylogenetic discontinuity represents introgression from one of the other Elephas spp. within the Asian branch itself.
The second hypothesis, that of persistence of two lineages through incomplete lineage sorting, has been cited as the probable cause for coexisting divergent haplotypes observed in African elephants (Georgiadis et al., 1994). Hartl et al. (1996), in a survey of cytochrome b variation in captive Asian elephants, observed eight haplotypes which clustered into two groups, which too was attributed to incomplete lineage sorting.
As a result of lineage sorting by random genetic drift, the maximum expected time of persistence of two selectively neutral haplotype lineages is 4Ne generations, where Ne is the effective population size of females (Kimura & Ohta, 1969). Thus, persistence of the two haplotype lineages observed in the present study through incomplete lineage sorting, necessitates a large long-term effective population size of females. Empirical evidence for large long-term effective population sizes is lacking even in species with current female census population sizes of many millions (Avise et al., 1988; Ramey, 1995). As the largest terrestrial mammals, elephants are unlikely to have had very large population sizes at any point in their evolution. In addition, low genetic diversity in Asian elephants observed in the present study and previous studies (Nozawa & Shotake, 1990; Hartl et al., 1995, 1996), the extinction of the majority of Proboscidean species that existed, and the palaeontological evidence for the expansion and contraction of species ranges, all point to the probability of large historical population fluctuations and a small long-term effective population size in Asian elephants. Thus, although incomplete lineage sorting cannot be excluded as the reason for the observed phylogeographic pattern, it seems an unlikely explanation.
The third hypothesis, that of allopatric divergence and secondary admixture, requires a concurring geological history. Although 20 miles of ocean currently separate Sri Lanka from the mainland, geological evidence indicates that they initially formed a single landmass (Jacob, 1949). A Pliocene subsidence of the northern part of Sri Lanka below sea level initially separated it from the mainland. Subsequent re-elevation of the land in the Pleistocene formed a land bridge, which repeatedly submerged and emerged with the rise and fall of sea levels caused by glaciation events (Jacob, 1949).
Thus it is conceivable that the two Asian elephant lineages observed in the present study evolved in Sri Lanka and the mainland during a long period of isolation and that gene flow occurred during more recent events of reconnection, giving rise to the present distribution of haplotypes.
As elephants have been domesticated for thousands of years and trade in elephants with importation and exportation of elephants from Sri Lanka has been recorded historically (Jayewardene, 1994), another possible cause of secondary admixture is movement of animals by humans. However, in the captive management of elephants, it is very rare for a female to be released or to escape into the wild. Such events are even less likely to have been allowed to occur historically, given the expense and effort that must have been incurred in transporting an elephant between the mainland and Sri Lanka at that time. In addition, although assemblage β was more widespread in Sri Lanka and assemblage α on the mainland, with nine and five haplotypes, respectively, three haplotypes of assemblage α and two haplotypes of assemblage β were also observed in Sri Lanka and the mainland, respectively. Although the possibility cannot be discounted, such an extent of admixture in both directions is unlikely to have occurred as a result of human intervention.
The two hypotheses of introgression of DNA from a closely related species and allopatric isolation with secondary admixture are not mutually exclusive. In fact, isolation and secondary admixture would be an integral part of a hypothesis of introgression from one of the other species within the Asian branch of the Elephas complex. Although the previous discussion presents a possible mechanism involving the mainland and Sri Lanka, a definitive conclusion awaits a broader analysis including samples from additional localities on the mainland, other continental islands such as Java and Sumatra and subfossil material from the progenitors of the Elephas lineage.
Genetic diversity
Our estimates of genetic diversity values in Asian elephants were lower than those estimated for the mitochondrial control region of many large mammals. Total nucleotide diversity in Asian elephants was 0.0176 compared to 0.029–0.100 for African ungulates (Simonsen et al., 1998), 0.0257 for humpback whales (Baker et al., 1993), and 0.040 for manatees (Garcia-Rodriguez et al., 1998). The total haplotype diversity value of 0.873 in Asian elephants was lower than values for African ungulates [0.99–0.95 (Simonsen et al., 1998)], but comparable to those of marine mammals [humpback whales 0.88 (Baker et al., 1993), and manatees 0.839 (Garcia-Rodriguez et al., 1998)]. Reported allozyme studies (Nozawa & Shotake, 1990; Hartl et al., 1995) and the study on cytochrome b variation (Hartl et al., 1996) in Asian elephants also found low levels of heterozygosity and thus corroborate the findings of the present study. Low levels of genetic variation can occur through founder events, bottlenecks and genetic drift, and are indicative of small long-term effective population sizes.
Although the genetic diversity within Asian elephants was low compared to other terrestrial large mammals, the levels observed do not indicate that Asian elephants are genetically depauperate. Allozyme studies in species such as the cheetah (O’Brien et al., 1986), and mitochondrial studies of mountain sheep in south-western United States and Mexico (Ramey, 1995) have shown much lower levels of genetic variability.
Although sample size in the present study was biased towards Sri Lanka and towards the mainland in Hartl et al.’s (1996) study, both studies found a higher level of genetic diversity in Sri Lanka. Hartl et al. (1996) assumed Asian elephant populations to be panmictic and attributed the lower heterozygosity on the mainland to a greater decrease of that population (to 5% of historical size) compared to Sri Lanka (to 20% of historical size). However, loss of heterozygosity subsequent to a permanent population decrease to an effective size Ne occurs on a time scale of 2Ne generations in a panmictic population (Wright, 1969). Thus, the decline in Asian elephant numbers is far too recent for detection of decreased heterozygosity. An assumption of population subdivision instead of panmixia can account for a short-term reduction in heterozygosity, because a decrease in range size of a subdivided population resembles sampling of a local population. However, it still cannot account for the greater reduction in heterozygosity on the mainland, as even the historical Sri Lankan range is but a fragment of the current mainland range.
In contrast, a hypothesis of allopatric divergence and secondary admixture provides sufficient explanation for the smaller population having greater genetic diversity, as gene flow from the larger mainland population to the smaller Sri Lankan population would have a larger impact on diversity indices in Sri Lanka, than the converse. Such an interpretation is supported by our finding of high frequencies of both assemblages in Sri Lanka and the predominance of a single assemblage on the mainland.
Molecular scatology
Molecular scatology is still an emerging field and holds much potential for the application of genetic analysis to free-ranging populations. The high degree of success (96%) in amplification of mtDNA from field-collected dung samples demonstrated in this study could result from physiological or physical attributes of the mode of digestion and diet of elephants. The collection of samples from the surface crust of dung boli could also be a factor in improving the success of amplification, as the surface would be the last in contact with the gut epithelium and first to dry upon deposition, and DNA contained therein would be the least degraded. The serial sampling experiment as well as reliable amplification of target sequences from samples estimated to be over two weeks old in this study, demonstrates that dung collected many days after deposition can provide DNA of sufficient integrity for analysis without special treatment, which greatly facilitates the collection of samples. The demonstrated ability to store samples at ambient temperature using simple reagents for long periods of time, simplifies the logistics of sample collection and enables significant reduction of sampling costs, facilitating the adoption of genetic analysis for large-scale surveys. Future developments in the field of molecular scatology will undoubtedly further facilitate genetic analysis of free-ranging populations and make significant contributions to the management and conservation of threatened species.
References
Arnason, U., Xu, X. and Gullberg, A. (1996). Comparison between the complete mitochondrial sequence of Homo and the common chimpanzee based on nonchimeric sequences. J Mol Evol, 42: 145–152.
Avise, J. C., Ball, R. M. and Arnold, J. (1988). Current versus historical population sizes in vertebrate species with high gene flow: a comparison based on mitochondrial DNA lineages and inbreeding theory for neutral mutations. Mol Biol Evol, 5: 331–344.
Baker, C. S., Perry, A., Bannister, J. L., Weinrich, M. T., Abernethy, R. B., Calambokidis, J. et al (1993). Abundant mitochondrial variation and world-wide population structure in humpback whales. Proc Natl Acad Sci USA, 90: 8239–8243.
Deraniyagala, P. E. P. (1955). Some Extinct Elephants, Their Relatives, and the Two Living Species. National Museum of Ceylon, Colombo, Sri Lanka.
Dowling, T. E., De-Marias, B. D., Minckley, W. L., Douglas, M. E. and Marsh, P. C. (1992). Use of genetic characters in conservation biology. Conserv Biol, 6: 7–8.
Drysdale, T. and Florkiewicz, R. F. (1989). Electrophoretic variation within and between the two extant elephant species (Mammalia: Proboscidea). J Mammal, 70: 381–383.
Fernando, P. (1998). Genetics, Ecology, and Conservation of the Asian Elephant. PhD Thesis, University of Oregon, Eugene, OR.
Garcia-Rodriguez, A. I., Bowen, B. W., Domning, D., Mignucci-Giannoni, A. A., Marmontel, M., Montoya-Ospina, R. A., et al. (1998). Phylogeography of the West Indian manatee (Trichechus manatus): how many populations and how many taxa? Mol Ecol, 7: 1137–1149.
Georgiadis, N., Bischoff, L., Templeton, A., Patton, J., Karesh, W. and Western, D. (1994). Structure and history of African elephant populations. I. Eastern and Southern Africa. J Hered, 85: 100–104.
Hartl, G. B., Kurt, F., Hemmer, W. and Nadlinger, K. (1995). Electrophoretic and chromosomal variation in captive Asian elephants (Elephas maximus). Zoo Biol, 14: 87–95.
Hartl, G. B., Kurt, F., Tiedemann, R., Gmeiner, C., Nadlinger, K., U-Mar, K. et al (1996). Population genetics and systematics of Asian elephant (Elephas maximus): a study based on sequence variation at the Cyt b gene of PCR-amplified mitochondrial DNA from hair bulbs. Zeitschrift fur Saugetierkunde, Int J Mam Biol, 61: 285–294.
Irwin, D. M., Kocher, T. D. and Wilson, A. C. (1991). Evolution of cytochrome b gene of mammals. J Mol Evol, 32: 128–144.
Jacob, K. (1949). Land connections between Ceylon and peninsular India. Proc Natl Inst Sci India, 15: 341–343.
Jayewardene, J. (1994). The Elephant in Sri Lanka Distributed by The Wildlife Heritage Trust of Sri Lanka, Colombo Sri Lanka.
Kimura, M. and Ohta, T. (1969). The average number of generations until fixation of a mutant gene in a finite population. Genetics, 61: 763–771.
Maglio, V. J. (1973). Origin and evolution of the Elephantidae. Trans Am Phil Soc Philadelphia, New Series, 63: 1–149.
Martin, A. P. and Palumbi, S. R. (1993). Body size, metabolic rate, generation time, and the molecular clock. Proc Natl Acad Sci USA, 90: 4087–4091.
Mckay, G. M. (1973). Behavior and ecology of the Asiatic elephant in southeastern Ceylon. Smithsonian Contrib Zool, 125: 1–113.
Nei, M. (1987). Molecular Evolutionary Genetics. Columbia University Press, New York.
Nozawa, K. and Shotake, T. (1990). Genetic differentiation among local populations of Asian elephant. Z Zool Syst Evol- Forsch, 28: 40–47.
O’brien, S. J., Wildt, D. E. and Bush, M. (1986). Can the cheetah survive? Sci Am, March,84–92.
Olivier, R. (1978). Distribution and status of the Asian elephant. Oryx, 14: 379–424.
Ramey II, R. R. (1995). Mitochondrial DNA variation, population structure, and evolution of mountain sheep in the south-western United States and Mexico. Mol Ecol, 4: 429–439.
Rozas, J. and Rozas, R. (1997). DnaSP, version 2.0: a novel software package for extensive molecular population genetic analysis. Computer Applic Biosci, 13: 307–311.
Santiapillai, C. and Jackson, P. (1990). The Asian Elephant: An Action Plan for its Conservation. IUCN/SSC Asian Elephant Specialist Group, IUCN, Gland.
Shoshani, J. and Eisenberg, J. F. (1982). Elephas maximus. Mammalian Species, 182: 1–8.
Simonsen, B. T., Siegismund, H. R. and Arctander, P. (1998). Population structure of African buffalo inferred from mtDNA sequences and microsatellite loci: High variation but low differentiation. Mol Ecol, 7: 225–237.
Sukumar, R. (1989). The Asian Elephant: Ecology and Management. Cambridge University Press, Cambridge.
Swofford, D. L. (1998). PAUP*Phylogenetic Analysis Using Parsimony (and Other Methods), version 4. Sinauer Associates, Sunderland, MA.
Wright, S. (1969). Evolution and the Genetics of Populations, vol. 2 The Theory of Gene Frequencies. University of Chicago Press, Chicago.
Zaykin, D. V. and Pudovkin, A. I. (1993). Two programs to estimate significance of χ2values using pseudo-probability tests. J Hered, 84: 15–15.
Acknowledgements
We thank L. K. A. Jayasinghe, H. K. Janaka, Manori Gunawardena, G. V. Gunewardene, Nimal Kaluarachchi, R. A. R. Perera, V. U. Weeratunga, H. Corea, M. Siriwardene, C. Gunasekere, N. Fernando, S. Udawatte, N. Dayawansa, and L. Seneviratne for field assistance in collecting samples from Sri Lanka, E. Wickramanayake for providing samples from Bhutan and Vietnam, R. Tizard for assistance in collecting samples from Laos, L. Egert, R. R. Ramey II and M. J. Schmidt for providing samples from zoo animals, and N. Lehman, T. Goldberg and K. Muralidharan for advice on molecular analysis. We also thank R. Rudran, J. H. Postlethwait, M. Lynch, J. Bonine and W. E. Bradshaw for commenting on an earlier version of the manuscript, and C. Wemmer for encouragement and providing the Asian elephant sequence. The Department of Wildlife Conservation, Sri Lanka, permitted collection of samples from protected areas. This work was supported in part by NSF grant DEB 9225127 to R. Lande and a WWF grant to P. Fernando.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fernando, P., Pfrender, M., Encalada, S. et al. Mitochondrial DNA variation, phylogeography and population structure of the Asian elephant. Heredity 84, 362–372 (2000). https://doi.org/10.1046/j.1365-2540.2000.00674.x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1046/j.1365-2540.2000.00674.x
Keywords
This article is cited by
-
Identification of conservation priority units in the Asian elephant, Elephas maximus
Conservation Genetics (2023)
-
Revisiting traditional SSR based methodologies available for elephant genetic studies
Scientific Reports (2021)
-
American mastodon mitochondrial genomes suggest multiple dispersal events in response to Pleistocene climate oscillations
Nature Communications (2020)
-
African elephant genetics: enigmas and anomalies\(^\dagger \)
Journal of Genetics (2019)
-
Generation of two-cell cloned embryos from mouse faecal cell
Scientific Reports (2018)
