
Abstract
Evolutionary Developmental Biology aims for a mechanistic understanding of phenotypic diversity, and present knowledge is largely based on gene expression and interaction patterns from a small number of well-known model organisms. However, our understanding of biological diversification depends on our ability to pinpoint the causes of natural variation at a micro-evolutionary level, and therefore requires the isolation of genetic and developmental variation in a controlled genetic background. The colour patterns of Heliconius butterflies (Nymphalidae: Heliconiinae) provide a rich suite of naturally occurring variants with striking phenotypic diversity and multiple taxonomic levels of variation. Diversification in the genus is well known for its dramatic colour-pattern divergence between races or closely related species, and for Müllerian mimicry convergence between distantly related species, providing a unique system to study the development basis of colour-pattern evolution. A long history of genetic studies has showed that pattern variation is based on allelic combinations at a surprisingly small number of loci, and recent developmental evidence suggests that pattern development in Heliconius is different from the eyespot determination of other butterflies. Fine-scale genetic mapping studies have shown that a shared toolkit of genes is used to produce both convergent and divergent phenotypes. These exciting results and the development of new genomic resources make Heliconius a very promising evo-devo model for the study of adaptive change.
Similar content being viewed by others

Heliconius butterflies: an emerging evo-devo model
Evolutionary developmental biology aims for a mechanistic understanding of the origins of phenotypic diversity. How are developmental pathways modified to produce evolutionary novelty? What are the genes that underlie evolutionary radiations or adaptive change? Do they share particular characteristics or modes of actions? The homeotic genes provide us with insights into major transitions in animal body plans and how genetic networks are modified in diverse taxa (Carroll et al, 2001; Davidson, 2001; Wilkins, 2002). In general, however, the evo-devo community has been slower to tackle the origins of recent evolutionary novelty (but see Bradshaw et al, 1998; Stern, 1998; Mundy et al, 2004; Colosimo et al, 2005; Gompel et al, 2005; Prud'homme et al, 2006). Such questions cannot be answered by studying a few distantly related model species. Instead, comparative studies are needed of closely related species that differ in traits of interest and require the development of new model systems that show great diversity among closely related species and forms but that are also amenable to evolutionary and ecological studies.
Butterfly wing patterns are excellent subjects for evo-devo studies because the patterns are structurally simple, highly variable, and, in many cases, the evolutionary and ecological significance of the pattern is well understood (see Brakefield et al, 1996; Beldade and Brakefield, 2002; McMillan et al, 2002; Beldade et al, 2005 for recent reviews). This is particularly true in the passion-vine butterflies, Heliconius (Nymphalidae: Heliconiinae), which combine extensive natural variation in colour pattern with a strong history of ecological and evolutionary research (eg, Benson, 1972; Gilbert, 1972; Brown, 1981; Mallet and Barton, 1989b; Joron et al, 1999; Jiggins et al, 2001; Kapan, 2001; Flanagan et al, 2004; Langham, 2004).
The group, composed of 40 species and hundreds of geographic variants across the Neotropics, shows significant variation in their wing patterns at every biological level from divergent species to sympatric colour morphs of the same species (Figure 1). The vivid colour patterns of Heliconius are adaptations that warn potential predators of the butterflies' unpalatability (Bates, 1862; Langham, 2004), presumably related to their evolutionary history in association with cyanogenic foodplants in the Passifloraceae (Brown, 1981; Engler et al, 2000). Nearly all Heliconius species participate in Müllerian mimicry association, such as the orange-rayed Amazonian mimicry ring which involves up to nine species of Heliconiines, or with other butterflies such as the ubiquitous tiger-striped Ithomiinae (Nymphalidae) in lowland and premontane forests of tropical America (Brown and Benson, 1974; Brown, 1981; Figure 1). Geographic radiation in Heliconius colour patterns closely follows the geography of mimicry in local butterfly communities, many species occurring as a mosaic of sharply defined races across Central and South America. Parapatric races hybridise in narrow hybrid zones stabilised by frequency-dependent selection which are permeable to gene flow across the genome except around colour-pattern loci (Mallet, 1993). Such colour-pattern hybrid zones are known to move across geographic areas, possibly due to positive selection for new mimicry patterns or alternatively driven by allelic dominance (Mallet and Barton, 1989a; Blum, 2002).

A sample of the morphological diversity of wing patterns in Heliconius and related genera. Each row represents a phylogenetic clade in the tribe Heliconiini. The phylogram on the left is a topology derived from mitochondrial and nuclear trees (Beltrán et al, 2002), highlighting the deeply diverged ‘melpomene’ (orange) and ‘erato’ (blue) clades of the genus. The phylogenetic position and consequent nomenclature of Neruda and Laparus are controversial and considered to fall within Heliconius on this figure (green). The figure highlights the rampant pattern diversification within clades and species, and mimicry between clades. Reconstruction of ancestral wing patterns is difficult for such rapidly evolving traits. It may be the case that the orange-rayed pattern is ancestral, in which case the other mimetic patterns are convergent derived patterns. However, it has also been suggested that ancestral Heliconius were more similar to the so-called ‘postman’ pattern of H. melpomene amaryllis, in which case the rayed pattern has evolved repeatedly.
Mimicry is common between members of diverged clades within Heliconius (Figure 1; note, for instance, the iridescent blue mimicry ring, or the black & white mimicry ring); in contrast sister species tend to diverge in pattern and mimicry associations, such as H. melpomene vs H. cydno, or H. burneyi vs H. wallacei (Figure 1; Beltrán, 2004). Indeed, Heliconius colour patterns are used as mating signals, and play an important role in speciation (McMillan et al, 1997; Jiggins et al, 2001; Kronforst et al, 2006). The radiation in Heliconius colour patterns thus couples both divergent evolution and multiple independent cases of convergent evolution representing of varying evolutionary timescales. The fact that these patterns have clear functional significance in nature will link developmental diversity, within-population adaptation, and macroevolution.
For this review, we concentrate on the role that heliconiine butterflies can play as an emerging evo-devo model of phenotypic change. Our aim is to provide (i) an overview of the extant diversity of wing patterns in the group, (ii) review what is known about the genetic basis of this diversity, and (iii) highlight emerging research, research directions, and research tools that promise to make Heliconius a model system for studying the interface between development and adaptive change.
The genetic architecture of pattern variation in Heliconius
The natural diversity of colour patterns found among Heliconius species and races is determined by adaptive combinations of alleles at a surprisingly reduced set of genetic loci of large phenotypic effect. These genes are most likely developmental genes that regulate the spatial expression of downstream scale maturation pathways, thus controlling the development of morphology and pigmentation of the future scales which generate adult wing patterns (Gilbert et al, 1988; Nijhout, 1991). Although the molecular nature of these genes is unknown, gene action and interactions are well characterised: many years of crossing experiments between species and races have shown how a handful of loci control phenotypic shifts across large areas of the wing surface, changing the position, size and shape of red/orange/yellow and melanic patches on both the dorsal and ventral surfaces of the fore and hindwings (H. numata: Brown and Benson, 1974; H. melpomene, H. erato: Sheppard et al, 1985; Mallet, 1989; H. erato/H. himera: Jiggins and McMillan, 1997; H. cydno: Kapan, 1998; H. cydno/H. melpomene: Gilbert, 2003; Naisbit et al, 2003).
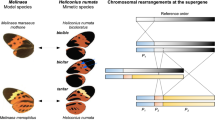
Alleles at major switch loci in Heliconius are natural variants, not laboratory generated mutants, and can be studied on a common genetic background by backcrossing pattern alleles between populations that are not genetically differentiated. Heliconius butterflies therefore offer an excellent opportunity to study the developmental and genetic basis of an adaptive radiation. Pattern variation is perhaps best understood in the two co-mimics H. erato and H. melpomene. The two species are distantly related, yet have undergone a parallel radiation into 23 colour pattern races (Turner, 1977). Although more than 20 different loci have been described in each radiation (Sheppard et al, 1985; Mallet, 1989; Jiggins and McMillan, 1997; Naisbit et al, 2003), geographic variation in wing pattern phenotype can be explained by allele changes at four to five loci of major effect. Pattern variation in H. numata provides perhaps one of the most striking examples of the broad action of major loci in Heliconius (Figure 2a and below). H. numata is closely related to H. melpomene and H. cydno (∼5% mtDNA divergence; Beltrán et al, 2002), but has evolved to mimic large and highly distasteful Ithomiines (Melinaea, Mechanitis) and Danaines (Lycorea). Its wings are characterised by patterns of black spots and stripes on an orange and yellow background, and all the local and geographic pattern variation maps to a single pattern locus known as ‘P’ (Brown and Benson, 1974; Joron, 2000). Hyperallelism is rampant and up to nine different alleles have been found to segregate in some populations (Figure 2b; Brown and Benson, 1974; Joron et al, 1999; Joron, 2000). The pattern of variation is therefore similar to the classic examples of polymorphism at colour-pattern ‘supergenes’ (clusters of tightly linked genes) in Batesian mimics such as Papilio dardanus and P. memnon (Turner, 1977).

The genetic basis of polymorphism in H. numata. Crosses between sympatric forms show inheritance of the whole colour pattern at the single Mendelian locus P, and a largely linear dominance series of the P alleles which avoids non-mimetic heterozygotes (Brown and Benson, 1974; Joron, 2000). (a) F2-type cross (B502) between Peruvian forms, showing the recessive allele silvana, and the segregation of three alleles into four discrete phenotypes in the progeny. Melanin patches are usually (although variably) dominant, and orange is always dominant over yellow. However, melanin patches may be recessive to orange or yellow scales, for instance the black hindwing margin and black forewing patch of the form silvana (PsilPsil), suggesting regulation of dominance selected for mimicry (Joron, 2000). A linkage map derived from this and other broods was generated as outlined in Joron et al (in press), and shows the position of the P locus (red bar) relative to flanking molecular markers. Recombination distances are in Haldane cM. These markers show positional homology between P and the colour pattern complex locus N/Yb/Sb in H. melpomene (Joron et al, in press), and are now being used for positional cloning. (b) Diversity of alleles found in some populations of north-eastern Peru. Form bicoloratus (allele Pbic) is the top dominant and silvana (Psil) the bottom recessive. Crosses and wild-caught recombinants suggest P to be a supergene (Brown and Benson, 1974), and some rare alleles such as lutea appear to be non-mimetic and may occur by recombination within the P locus. Allelic differences at the P locus are selected for mimicry of local Melinaea and Mechanitis butterflies (Nymphalidae: Ithomiinae). The single-locus inheritance of pattern in H. numata is likely a result of the atypical selection pressures associated with multiple mimicry (Brown and Benson, 1974; Joron et al, 1999).
Initially, the observation that adaptive variation in wing patterns in Heliconius was the result of a small number of major ‘switch’ loci was thought to be an unusual artefact of Müllerian mimicry selection, where the adaptive landscape was envisioned to be more rugged than that for most adaptive traits (Turner, 1988; Mallet, 1993; Coyne et al, 1997). However, although multiple loci are known to control morphologies under strong selection, such as some of the domestication traits in maize (Westerbergh and Doebley, 2002; Doebley, 2004), a growing number of studies on organisms ranging from plants to fish (eg monkeyflowers: Bradshaw et al, 1998; sticklebacks: Cresko et al, 2004; Colosimo et al, 2005) have shown that a small number of loci with large phenotypic effect often underlie adaptation, suggesting that the architecture of phenotypic evolution in Heliconius may be more typical of adaptive change than previously realised.
Macroevolutionary importance of Heliconius patterns
The genes that control wing patterns in Heliconius appear to be preserved across species boundaries. Reproductive barriers are often incomplete between closely related species, permitting interspecific crosses (Jiggins and McMillan, 1997; Gilbert, 2003; Naisbit et al, 2003). These studies demonstrate that colour-pattern differences between closely related species appear to be caused by allelic differences at same loci that are responsible for phenotypic differences within a species (Table 1). This is true even when speciation is coupled with a shift in mimetic alliance (Jiggins and McMillan, 1997; Naisbit et al, 2003), which occurs commonly in Heliconius (Beltrán, 2004). In combination with visual mate searching using wing colour signals, large shifts in pattern can lead to speciation (McMillan et al, 1997; Jiggins et al, 2001; Naisbit et al, 2001). Thus, there is a direct link between phenotypic shifts in pattern caused by just a few genetic changes and macroevolutionary diversification.
Rules and constraints on pattern formation
The radiation in the mimetic wing patterns of Heliconius butterflies provides an excellent model system for exposing the nature of constraints, bias, optimality, and chance in morphological change. Several authors have attempted to draw generalisations from the wealth of crossing data in Heliconius (eg, Turner, 1977; Sheppard et al, 1985). Notably Gilbert (2003), synthesising nearly 30 years of his own crossing work, identified a number of predictable dominance/epistatic effects in his crosses and defined three scale types based on pigment type and scale morphology (Gilbert et al, 1988; Gilbert, 2003). Type I scales are white (pigmentless) or yellow (3-hydroxy-L-kynurenine); whereas Type II scales are black (melanin) and Type III scales are brown/red/orange (xanthommatin and di-hydro-xanthommatin). Alleles controlling type III scales are generally dominant to those controlling Type II scales, which are dominant to Type I scales. These patterns of dominance can also apply to inter-locus epistasis where two different loci influence the same wing region. Nonetheless, although generally true in H. melpomene and H. erato these generalisations do not apply to all Heliconius. For example, in H. numata melanin patterns are usually dominant over both orange and yellow (ie Type I<Type III<Type II), and in some cases yellow elements are even dominant to melanic ones, a complete reversal of the typical scheme (see Figure 2; Joron, 2000). Clearly, selection for mimicry can and does break the ‘rules’, highlighting the flexibility of butterfly wing patterns (Beldade et al, 2002b).
It has also been suggested that a common developmental process might constrain, or bias pattern evolution in Heliconius. At one extreme, it has been hypothesised that the precise and repeated convergent evolution between the two relatively distantly related co-mimics, H. erato and H. melpomene, is caused by changes at homologous loci (Turner, 1984; Nijhout, 1991). However, there are notable differences in the exact nature of the phenotypes and their genetic control that has led others to argue that there is little homology between the species (Mallet, 1989). As mentioned above, the same pigment biosynthesis pathways are involved in both species, so questions about homology essentially relate to pattern formation prior to pigment production. However, the two species do not interbreed and the question of homology of genetic control between the two mimics is only now being addressed with molecular markers.
From patterns to genes
Application of molecular markers to crossing experiments
High-resolution linkage maps and the development of molecular markers transferable between species are allowing researchers to explore the architecture of convergent and divergent evolution in Heliconius and to test hypotheses about genetic homology between mimetic species. Mapping work in Heliconius takes advantage of the fact that controlled crosses between divergent colour pattern forms can be designed to follow the segregation of specific colour pattern alleles (Figures 2 and 3). This forward genetic approach has coupled Amplified Fragment Length Polymorphisms (AFLP) fingerprints with co-dominant anchor loci. AFLPs are a powerful technique for unexplored genomes (Mueller and Wolfenbarger, 1999; Parsons and Shaw, 2002) and, in Heliconius have allowed researchers to quickly home in on the regions of the genome that contain major colour pattern genes (Jiggins et al, 2005; Tobler et al, 2005; Kapan et al, in press). Indeed, there is now a tight association between AFLP markers and several major colour pattern loci in both H. erato and H. melpomene. For example, the N/Yb/Sb gene complex in H. melpomene and the D and the Sd locus in H. erato have all been localised and a number of tightly linked AFLP bands isolated (Jiggins et al, 2005; Kapan et al, in press; Figure 3). AFLP bands of interest can then be isolated, cloned and sequenced, and converted into co-dominant loci by designing primers that specifically amplify the AFLP fragment of interest (Beltrán, 2004). These ‘targeted’ AFLP loci work across different mapping families, facilitating finer precision mapping and are also an excellent source of probes for BAC libraries, which are now available for H. erato, H. melpomene, and H. numata.

Segregation of two patterning loci in a single cross and their genomic position in H. erato. Segregating variation in a cross between H. erato etylus (rayed parent) and H. himera. The F1 individuals of this cross lack the forewing yellow band, possess an orange patch on the proximal part of the forewing and show both the characteristic rays and hindwing bar of the parental species. The effects of two major co-dominant loci segregating can be seen in F2 offspring (Punnet square). Linkage maps of the two colour pattern linkage groups, LG03 and LG06, were generated as outlined in Kapan et al (in press). The blue stars represent AFLP markers on each chromosome. The red bar represents the interval across each chromosome where the colour pattern locus can be placed with high confidence. Bulk segregant analysis has generated a dense cluster of AFLP markers tightly linked to each of the two colour pattern loci, which are now being used for positional cloning.
In addition to AFLP markers targeted to colour pattern genes, the linkage maps are increasingly utilising co-dominant loci, which include microsatellites (Flanagan et al, 2002; Mavárez and González, 2006) and single copy nuclear loci (Beltrán et al, 2002; Kronforst, 2005; Papanicolaou et al, 2005), useful for anchoring and comparing maps from different crosses or species. In particular, a number of ‘candidate’ genes, chosen based on knowledge of gene action in other organisms, have now been mapped relative to the loci that cause pattern change in Heliconius. Candidate gene approach has been very successful in other organisms. For example, expression studies for genes known to be involved in Drosophila wing development revealed novel but related roles of such genes in pattern specification and variation in butterflies (Carroll et al, 1994; Brakefield et al, 1996; Brunetti et al, 2001; Beldade et al, 2002a). In Heliconius, however, this approach has allowed us to rule out most potential candidates by linkage mapping (Jiggins et al, 2005; Joron et al, in press; Kapan et al, in press). With the notable exception of tight linkage between wingless and the white/yellow colour switch locus K in H. cydno (Kronforst et al, 2006), several loci that are important in Drosophila wing development (apterous, wingless), in Bicyclus eyespot specification (distal-less, hedgehog, patched, cubitus interruptus), or in scale pigment synthesis (vermilion, cinnabar), are unlinked to pattern switch genes in one or more Heliconius species (see eg Jiggins et al, 2005; Tobler et al, 2005; Kapan et al, in press). These results can be disheartening, but they may imply, together with expression studies (Reed and Nagy, 2005), that novel or unexpected genes or pathways are involved in pattern specification.
The growing number of co-dominant markers that have been mapped in several Heliconius species also allows comparisons of gene order between species. Results so far indicate that gene order is well conserved across Heliconius (Joron et al, in press; Kapan et al, in press). Indeed, to date no conflicting linkage relationship has been found between H. erato, H. melpomene, and H. numata. The strongest support for the general conservation of linkage relationships within Heliconius comes from a cluster of ribosomal proteins (RpL5, RpS5, RpL10a, RpS8, RpP0), all of which map to the same linkage group and show conserved gene order in the three species (Jiggins et al, 2005; Joron et al, in press; Kapan et al, in press). Interestingly, the ribosomal cluster spans 47 cM in H. melpomene vs 32 cM in H. erato, which contrasts with the differences in genome size estimates, H. erato's genome being 36% larger than H. melpomene's (Jiggins et al, 2005). The apparently higher crossing-over frequency in H. melpomene vs H. erato is intriguing in light of evidence that H. erato may commonly act as the Müllerian model in this pair (Mallet, 1999; Flanagan et al, 2004). This suggests the interesting hypothesis that the genomic flexibility provided by increased recombination rate could help H. melpomene track pattern variation in H. erato. Efforts are now underway to develop and map many more anchor loci. Generating new anchor loci is now extremely efficient using EST sequences, with a primer design success rate of ca. 80% (ie 80% of primer pairs designed work in all three species; Papanicolaou et al, 2005). Furthermore, assigning loci to particular linkage groups is easy in Lepidoptera, because there is no crossing over during oogenesis (Suomalainen et al, 1973), and chromosomes are therefore inherited from the mother without recombination.
The linkage maps available for several Heliconius species and the high degree of conservation of gene order now allows a direct comparison of the genetic architecture underlying pattern evolution and a test of the long-standing hypothesis that homologous genes might be controlling mimcry in Heliconius. This comparative approach is yielding exciting results. Using a combination of ‘targeted’ AFLP markers and gene-based markers developed from initial genomic sequence, it has recently been demonstrated that N/Yb/Sb complex in H. melpomene, the Cr locus in H. erato, and the P locus in H. numata, all map to homologous 1 cM regions of the genome (Figure 2; Joron et al, in press). The N/Yb/Sb complex of H. melpomene and the Cr locus of H. erato similarly affect the distribution of white and yellow patches on the fore and hind wing of both radiations and is one of four or five patterning loci that underlie convergent change in the wing patterns of the co-mimics (see Mallet, 1989; Jiggins et al, 2005; Kapan et al, in press). The observation that loci with similar phenotypic effects map to the same genomic region is the first direct support for the hypothesis that homologous genes or complex of genes regulate convergence in the two co-mimics. Additional, albeit less compelling, support for this hypothesis comes from emerging patterns of linkage at other major color pattern loci in the two radiations, such as the D locus in H. erato and the D/B complex in H. melpomene which have nearly identical phenotypic effects on red patterning in the two co-mimics and both map to the same end of homologous linkage groups (Joron et al, in press; Kronforst et al, unpublished). Similarly, the Sd locus in H. erato and the Ac locus in H. cydno and H. melpomene, both of which effect the pattern of melanic scales on the forewing, also map to the same linkage group (Kronforst et al, unpublished).
The observation that mimicry between H. erato and H. melpomene appears to be regulated by many of the same loci or suite of loci, as predicted by Nijhout (1991), might suggest that some developmental constraints are important in mimetic evolution in Heliconius. However, this sharply contrasts with the lack of similarity between the patterns of the H. erato–H. melpomene pair and those of H. numata. Despite strong evidence for genetic homology, there is no obvious phenotypic homology between the effects of allelic substitutions at P in H. numata and N/Yb/Sb in H. melpomene. The P locus regulates pattern diversity much more broadly by affecting the distribution of yellow, brown/orange, and black color patterns elements across the whole wing surface (Figure 2). Rather than constraints, these observations therefore demonstrate an extraordinary flexibility of homologous color pattern genes in Heliconius, which respond to a variety of selection pressures for mimicry of divergent color patterns. This conserved locus, or ‘developmental hotspot’ (sensu Richardson and Brakefield, 2003), is responsible for pattern variation in at least three species belonging to two diverged clades, and appears to play a disproportionate role in both divergent and convergent adaptive evolution in the genus. The architecture, identity, and mode of action of this genomic region remain to be characterised to better understand its role and flexibility.
Prospects for positional cloning of pattern genes
Of course, much of the mapping work in Heliconius is directed towards identifying the loci that control pattern variation. The increase in genomic resources including (1) large numbers of replicate crosses, (2) ‘targeted’ loci near colour pattern genes and (3) BAC libraries make this a realistic goal and one that is achievable within the next few years. Moving forward requires linking the recombination maps to physical maps of the corresponding region. In H. melpomene the AFLP marker linked to the Yb gene has been used to screen a BAC library and construct a 500 kb contig tightly linked to the patterning genes. Segregating variation at BAC end sequences in the broods has been used to determine the direction in which Yb lies and locate recombination breakpoints between the pattern genes and flanking markers. Candidate patterning genes will be found by sequencing the region within these physical boundaries and locating the open reading frames (ORF). Markers developed in H. melpomene from BAC sequences are simultaneously being used to narrow down the region containing the hypothesised homologues P in H. numata and Cr in H. erato in the same way. Having exhausted the mapping resolution of crosses in each species, the involvement of the gene(s) identified from this region in colour pattern variation will be tested using various methods: expression studies on developing wings (eg Reed and Nagy, 2005) can test the up- or downregulation of genes identified from the BAC sequences, while association studies using BAC-derived markers in wild populations (eg Colosimo et al, 2005) can take advantage of historical recombination around the colour-pattern locus to identify narrower regions associated to specific genotypes. More targeted reverse genetics methods aimed at disrupting or enhancing specific gene expression, such as germline transformation (Peloquin et al, 2000; Marcus et al, 2004) and especially RNA interference (Fabrick et al, 2004; Eleftherianos et al, in press), have been successfully applied to lepidopteran species. Such techniques become increasingly transferable to diverse species (Marcus, 2005), and represent a promising way to test the involvement of genes in wing pattern phenotypes for species with rapid development such as Heliconius. The isolation of pattern genes is likely to be of major importance in our understanding of how the regulation at single loci can produce very different phenotypes. The comparative architecture and micro-synteny of targeted regions of the genome, such as that containing the complex loci P or N/Yb/Sb, will also tell us about the evolution of recombination patterns around genes under selection.
Indeed, the data already offer insights into how the ‘supergene’ in H. numata might have evolved. The pattern seen in H. melpomene and H. erato is one of geographic variation largely controlled by three to four clusters of tightly linked elements found on different chromosomes. One of these elements has taken over control of the entire pattern in H. numata, presumably facilitated by the fact that these regions already have major phenotypic effects on different parts of the wing in the ancestral species. There is also the possibility that the linked elements Yb, Sb and N found in H. melpomene might have been brought closer together to reduce the production of unfit intermediate genotypes in polymorphic H. numata populations. While the evolution of linkage between unlinked loci seems unlikely on theoretical grounds (Charlesworth and Charlesworth, 1975), a gradual reduction in recombination between already tightly linked elements seems more plausible. Thus, evolution of the H. numata supergene could have involved elements of both the ‘macromutationist’ and the ‘gradualist’ positions in this historical debate. Characterisation of the molecular basis of these genes will allow a direct test of these ideas.
From genes to pathways
From an evo-devo perspective, the major interest lies in linking the loci underlying pattern change in Heliconius, the so-called switch genes, with the pathways involved in wing pattern development. Identifying the pathways or modules of genes involved in wing pattern development promises to open an entirely new set of questions. For instance, in cases of convergence, we can determine (i) the level in pathways where changes tend to occur, (ii) if certain types of molecules (signalling molecules, transcription factors, pigment enzymes, transporters, etc.) play a disproportionate role, and (iii) the nature of constraint and potential in different pathway elements for producing similar phenotypes.
Pattern formation on butterfly wings is envisioned to be a multistep process. In early development, genes involved in cell-signalling and signal transduction work together to ‘pre-pattern’ or specify the fate (colour and morphology) of the individual scale cells that pattern the wing. The final adult colour pattern is produced later in development when scale cells interpret this positional information and produce pattern-specific colour pigments (Nijhout, 1991). Work on the early stages of pattern formation in Heliconius (Reed and Serfas, 2004; Reed and Nagy, 2005) has not, as yet, yielded the striking association between gene expression patterns and wing pattern elements seen in the eyespots of other butterflies (Carroll et al, 1994; Brakefield et al, 1996; Brunetti et al, 2001). Nonetheless, these studies suggest that at least part of the difference in patterning between butterfly groups may be due to temporal changes in conserved pattern-formation processes. For instance, the Notch/Distal-less (N/Dll) pattern formation process, associated with intervein elements and particularly eyespots in Bicyclus or Junonia, is truncated in Heliconius and other species lacking eyespots (Reed and Serfas, 2004). This implies that pattern variation within Heliconiines might be associated with the regulation of earlier stages of the N/Dll pattern-formation process, or possibly involve distinct pathways altogether. Actually, the apparent differences in the developmental architecture between eyespots and the coloured bands of Heliconius are not entirely unexpected. Eyespots are highly localised pattern elements relative to the large patterns of Heliconius.
The recent discovery of a mutant H. cydno with greatly reduced wing veins further highlights the differences between eyespots and Heliconius patterns. This veinless mutant had a pattern that was very similar to the wild-type, implying that, unlike eyespots, Heliconius patterns develop independently of wing veins (Reed and Gilbert, 2004). The vein independence of the Heliconius patterns also seems to disprove Nijhout's (1991) hypothesis of a common ‘nymphalid ground plan’ in which the Heliconius patterns represent an expansion of vein-dependent pattern elements found in other nymphalid butterflies. The Heliconius patterns more probably result from a distinct, and unexplored, whole-wing proximodistal patterning system established in the larval wing disc (Reed and Gilbert, 2004).
Beyond candidate genes
The candidate gene approaches described above are largely based on inferences about gene actions and interactions gleaned from research on Drosophila. This research avenue has clearly yielded insights into the mechanism of pattern formation on butterfly wings. However, the development of scale-covered wings and the patterning system for pigmenting them are evolutionary innovations of the Lepidoptera (Nijhout, 1991) and must therefore involve either novel genes or novel functions for conserved genes. To identify genes expressed during wing formation, sequencing projects are currently underway in both H. erato and H. melpomene. We have a growing database of Expressed Sequence Tags (ESTs), the bulk of which come from wing disc cDNA libraries (Papanicolaou et al, 2005). To date, approximately 10 000 Heliconius ESTs have been clustered and annotated with hierarchical BLAST searches, putative protein translations, and gene ontology (GO) terms, and are publicly available at www.heliconius.org. ESTs are important source of loci known to be expressed during wing pattern development in Heliconius and this ‘first-pass’ has identified key members of both cell signalling and pigment synthesis pathways (Table 2; Papanicolaou, 2005). In addition, EST sequences are an important source of new PCR-based markers for linkage mapping and will allow AFLP maps to be compared both within and between Heliconius species (Papanicolaou, 2005; Joron et al, in press).
A significant step towards understanding the sequence of gene expression during wing development is now possible by turning the emerging EST data into a gene chip (DNA microarray) to use during the development of Heliconius developing wings. The wing discs of Heliconius are large and completely accessible to sampling throughout development. Furthermore, they do not undergo complex morphogenetic movements (like the evagination of Drosophila wing disks), and the shape and pattern of the developing wing imaginal disk maps directly onto that of the adult wing. These factors facilitate developmental research in pattern formation that, when coupled with the large natural variation in Heliconius wing patterns, promise insights into how gene expression varies between (i) different parts of the developing wing, (ii) different geographic variants of the same species, and (iii) different species with convergent morphologies. These data will form the foundation for uncovering the networks that connect patterning genes to the pigment synthetic pathways, and how these networks change during pattern evolution. At the most basic level, these data promises a new suite of candidate loci, whose expression patterns can be tested using quantitative reverse-transcriptase PCR and in situ hybridisation (following eg Reed and Nagy, 2005) and whose position relative to the known ‘mimicry’ genes can be readily mapped. Furthermore, since eyespot specification is shared by most Nymphalids and may have been lost or truncated in Heliconius (Reed and Serfas, 2004), and since various kinds of banding patterns are also common in many Nymphalids including Bicyclus, the developmental underpinning of the bold patterns of Heliconius will provide an interesting contrast to the formation of eyespots in Junonia and Bicyclus (Brakefield et al, 1996; McMillan et al, 2002; Beldade et al, 2002a; Monteiro et al, 2003).
Perspectives
Advances in genomic resources, including high-resolution maps, BAC libraries, EST scans, and gene chips, are now offering exciting possibilities for comprehensive analyses of colour pattern change in Heliconius. So far, research has focussed on a trio of species encompassing most aspects of colour pattern evolution including geographic diversification (in each of the three species), local polymorphism (H. numata), diverging mimetic associations between closely related species (H. melpomene vs H. numata) as well as convergent phenotypes between distantly related species (H. erato vs H. melpomene), and makes Heliconius an excellent model for comprehensive analyses of colour pattern evo-devo. One of the advantages of the Heliconius system is the potential for direct identification of pattern switch genes by positional cloning. This approach avoids any dependence on prior identification of candidate genes and promises to uncover the genes responsible for the N/Yb/Sb complex of H. melpomene, the P locus in H. numata, and the Cr locus in H. erato in the near future. The evidence for positional homology between pattern switch genes within Heliconius offers exciting possibilities for comparative studies of the pattern specification pathways and insights into to the evolution of genome complexity, synteny, recombination rates, and cis-regulatory change.
As new candidate loci emerge the challenge will be to carry out the experimental studies that will provide a more detailed picture of the networks that connect the switch genes of Heliconius to pigment synthesis pathways, and how these networks change during adaptive radiation. On a broader phylogenetic scale, a general mechanistic understanding of wing pattern formation will require an appreciation of the interplay of different patterning systems on the developing wing, such as eyespot and banding patterns. Integrating our knowledge of several kinds of pattern specification will permit a fuller understanding of pattern evolution and how developmental processes are shaped by selective pressures.
References
Bates HW (1862). Contributions to an insect fauna of the Amazon valley. Lepidoptera: Heliconidae. Trans Linn Soc London 23: 495–566.
Beldade P, Brakefield PM (2002). The genetics and evo-devo of butterfly wing patterns. Nat Rev Genet 3: 442–452.
Beldade P, Brakefield PM, Long AD (2002a). Contribution of Distal-less to quantitative variation in butterfly eyespots. Nature 415: 315–318.
Beldade P, Brakefield PM, Long AD (2005). Generating phenotypic variation: prospects from ‘evo-devo’ research on Bicyclus anynana wing patterns. Evol Dev 7: 101–107.
Beldade P, Koops K, Brakefield PM (2002b). Developmental constraints versus flexibility in morphological evolution. Nature 416: 844–847.
Beltrán M (2004). The speciation history of Heliconius: inferences from multilocus DNA sequence data. Ph.D. diss., University College London, UK.
Beltrán M, Jiggins CD, Bull V, Linares M, McMillan WO, Bermingham E et al (2002). Phylogenetic discordance at the species boundary: comparative gene genealogies among rapidly radiating Heliconius butterflies. Mol Biol Evol 19: 2176–2190.
Benson WW (1972). Natural selection for Müllerian mimicry in Heliconius erato in Costa Rica. Science 176: 936–939.
Blum MJ (2002). Rapid movement of a Heliconius hybrid zone: evidence for phase III of Wright's shifting balance theory? Evolution 56: 1992–1998.
Bradshaw HD, Otto J, Frewen BE, McKay JK, Schemske DW (1998). Quantitative trait loci affecting differences in floral morphology between two species of monkeyflower (Mimulus). Genetics 149: 367–382.
Brakefield PM, Gates J, Keys DN, Kesbeke F, Wijngaarden PJ, Monteiro A et al (1996). Development, plasticity and evolution of butterfly eyespot patterns. Nature 384: 236–242.
Brown KS (1981). The biology of Heliconius butterflies and related genera. Annu Rev Entomol 26: 427–456.
Brown KS, Benson WW (1974). Adaptive polymorphism associated with multiple Müllerian mimicry in Heliconius numata. Biotropica 6: 205–228.
Brunetti CR, Selegue JE, Monteiro A, French V, Brakefield PM, Carroll SB (2001). The generation and diversification of butterfly eyespot color patterns. Curr Biol 11: 1578–1585.
Carroll SB, Gates J, Keys DN, Paddock SW, Panganiban GEF, Selegue JE et al (1994). Pattern formation and eyespot determination in butterfly wings. Science 265: 109–114.
Carroll SB, Grenier JK, Weatherbee SD (2001). From DNA to diversity: Molecular genetics and the evolution of animal design. Blackwell Science: Oxford.
Charlesworth D, Charlesworth B (1975). Theoretical genetics of Batesian mimicry. II. Evolution of supergenes. J Theor Biol 55: 305–324.
Colosimo PF, Hosemann KE, Balabhadra S, Villarreal G, Dickson M, Grimwood J et al (2005). Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science 307: 1928–1933.
Coyne JA, Barton NH, Turelli M (1997). Perspective: a critique of Sewall Wright's shifting balance theory of evolution. Evolution 51: 643–671.
Cresko WA, Amores A, Wilson C, Murphy J, Currey M, Phillips P et al (2004). Parallel genetic basis for repeated evolution of armor loss in Alaskan threespine stickleback populations. Proc Natl Acad Sci USA 101: 6050–6055.
Davidson EH (2001). Genomic Regulatory Systems: Development and Evolution. Academic Press: San Diego, CA.
Doebley J (2004). The genetics of maize evolution. Annu Rev Genet 38: 37–59.
Eleftherianos IPJM, Ffrench-Constant R, Reynolds SE (in press). RNAi suppression of recognition protein mediated immune responses in the tobacco hornworm Manduca sexta causes increased susceptibility to the insect pathogen Photorhabdus. Dev Comp Immunol (doi:10.1016/j.dci.2006.02.008).
Engler HS, Spencer KC, Gilbert LE (2000). Preventing cyanide release from leaves. Nature 406: 144–145.
Fabrick JA, Kanost MR, Baker JE (2004). RNAi-induced silencing of embryonic tryptophan oxygenase in the Pyralid moth, Plodia interpunctella. J Insect Sci 4: 15.
Flanagan N, Tobler A, Davison A, Pybus OG, Kapan DD, Planas S et al (2004). The historical demography of Müllerian mimicry in the Neotropical Heliconius butterflies. Proc Natl Acad Sci USA 101: 9704–9709.
Flanagan NS, Blum MJ, Davison A, Alamo M, Albarran R, Faulhaber K et al (2002). Characterization of microsatellite loci in neotropical Heliconius butterflies. Mol Ecol Notes 2: 398–401.
Gilbert LE (1972). Pollen feeding and reproductive biology of Heliconius butterflies. Proc Natl Acad Sci USA 69: 1403–1407.
Gilbert LE (2003). Adaptive novelty through introgression in Heliconius wing patterns: evidence for a shared genetic ‘tool box’ from synthetic hybrid zones and a theory of diversification. In: Boggs CL, Watt WB, Ehrlich PR (eds) Ecology and evolution taking flight: Butterflies as model systems. University of Chicago Press: Chicago. pp Ch. IV: 14.
Gilbert LE, Forrest HS, Schultz TD, Harvey DJ (1988). Correlations of ultrastructure and pigmentation suggest how genes control development of wing scales of Heliconius butterflies. J Res Lepid 26: 141–160.
Gompel N, Prud'homme B, Wittkopp PJ, Kassner VA, Carroll SB (2005). Chance caught on the wing: cis-regulatory evolution and the origin of pigment patterns in Drosophila. Nature 433: 481–487.
Jiggins CD, McMillan WO (1997). The genetic basis of an adaptive radiation: warning colour in two Heliconius species. Proc R Soc London Ser B 264: 1167–1175.
Jiggins CD, Mavárez J, Beltrán M, McMillan WO, Johnston JS, Bermingham EB (2005). A genetic linkage map of the mimetic butterfly, Heliconius melpomene. Genetics 171: 557–570.
Jiggins CD, Naisbit RE, Coe RL, Mallet J (2001). Reproductive isolation caused by colour pattern mimicry. Nature 411: 302–305.
Joron M (2000). Warning colour and Müllerian mimicry: the puzzle of diversification. Ph.D. diss., Université de Montpellier, France.
Joron M, Papa R, Beltrán M, Chamberlain N, Mavárez J, Baxter SW et al (in press). A conserved supergene locus controls colour pattern diversity in Heliconius butterflies. PLoS Biol.
Joron M, Wynne IR, Lamas G, Mallet J (1999). Variable selection and the coexistence of multiple mimetic forms of the butterfly Heliconius numata. Evol Ecol 13: 721–754.
Kapan DD (1998). Divergent natural selection and Müllerian mimicry in polymorphic Heliconius cydno (Lepidoptera: Nymphalidae). Ph.D. diss., University of British Columbia, Vancouver, Canada.
Kapan DD (2001). Three-butterfly system provides a field test of Müllerian mimicry. Nature 409: 338–340.
Kapan DD, Flanagan NS, Tobler A, Papa R, Reed RD, Acevedo González J et al (in press). Localization of Müllerian mimicry genes on a dense linkage map of Heliconius erato. Genetics (doi:10.1534/genetics.106.057166).
Kronforst MR (2005). Primers for the amplification of nuclear introns in Heliconius butterflies. Mol Ecol Notes 5: 158–162.
Kronforst MR, Young LG, Kapan DD, McNeely C, O'Neill RJ, Gilbert LE (2006). Linkage of butterfly mate preference and wing color preference cue at the genomic location of wingless. Proc Natl Acad Sci USA 103: 6575–6580.
Langham GM (2004). Specialized avian predators repeatedly attack novel color morphs of Heliconius butterflies. Evolution 58: 2783–2787.
Mallet J (1989). The genetics of warning colour in Peruvian hybrid zones of Heliconius erato and H. melpomene. Proc R Soc London Ser B 236: 163–185.
Mallet J (1993). Speciation, raciation, and color pattern evolution in Heliconius butterflies: Evidence from hybrid zones. In: Harrisson RG (ed) Hybrid zones and the evolutionary process. Oxford University Press: New York. pp 226–260.
Mallet J (1999). Causes and consequences of a lack of coevolution in Müllerian mimicry. Evol Ecol 13: 777–806.
Mallet J, Barton N (1989a). Inference from clines stabilized by frequency-dependent selection. Genetics 122: 967–976.
Mallet J, Barton NH (1989b). Strong natural selection in a warning colour hybrid zone. Evolution 43: 421–431.
Marcus JM (2005). Jumping genes and AFLP maps: transforming lepidopteran color pattern genetics. Evol Dev 7: 108–114.
Marcus JM, Ramos DM, Monteiro A (2004). Germline transformation of the butterfly Bicyclus anynana. Proc R Soc London B 271: S263–S265.
Mavárez J, González M (2006). A set of microsatellite markers for Heliconius melpomene and closely related species. Mol Ecol Notes 6: 20–23.
McMillan WO, Jiggins CD, Mallet J (1997). What initiates speciation in passion-vine butterflies? Proc Natl Acad Sci USA 94: 845–851.
McMillan WO, Monteiro A, Kapan DD (2002). Development and evolution on the wing. Trends Ecol Evol 17: 125–133.
Monteiro A, Prijs J, Bax M, Hakkaart T, Brakefield PM (2003). Mutants highlight the modular control of butterfly eyespot patterns. Evol Dev 5: 185–187.
Mueller UG, Wolfenbarger LL (1999). AFLP genotyping and fingerprinting. Trends Ecol Evol 14: 389–394.
Mundy NI, Badcock NS, Hart T, Scribner K, Janssen K, Nadeau NJ (2004). Conserved genetic basis of a quantitative plumage trait involved in mate choice. Science 303: 1870–1873.
Naisbit RE, Jiggins CD, Mallet J (2001). Disruptive sexual selection against hybrids contributes to speciation between Heliconius cydno and H. melpomene. Proc R Soc London Ser B 268: 1849–1854.
Naisbit RE, Jiggins CD, Mallet J (2003). Mimicry: developmental genes that contribute to speciation. Evol Dev 5: 269–280.
Nijhout HF (1991). The development and evolution of butterfly wing patterns. In: Funk VA, Cannell PF (eds) Smithsonian Series in Comparative Evolutionary Biology. Smithsonian Institution Press: Washington, DC.
Papanicolaou A (2005). Comparative genomics and genetic linkage mapping between the neotropical butterflies, Heliconius melpomene (Linnaeus) and Heliconius erato (Linnaeus). Masters of Science by Research diss., University of Edinburgh, Edinburgh.
Papanicolaou A, Joron M, McMillan WO, Blaxter ML, Jiggins CD (2005). Genomic tools and cDNA derived markers for butterflies. Mol Ecol 14: 2883–2897.
Parsons YM, Shaw KL (2002). Mapping unexplored genomes: a genetic linkage map of the Hawaiian cricket Laupala. Genetics 162: 1275–1282.
Peloquin JJ, Thibault ST, Staten R, Miller TA (2000). Germ-line transformation of pink bollworm (Lepidoptera: Gelechiidae) mediated by the piggyBac transposable element. Insect Mol Biol 9: 323–333.
Prud'homme B, Gompel N, Rokas A, Kassner VA, Williams TM, Yeh SD et al (2006). Repeated morphological evolution through cis-regulatory changes in a pleiotropic gene. Nature 440: 1050–1053.
Reed RD, Gilbert LE (2004). Wing venation and Distal-less expression in Heliconius butterfly wing pattern development. Dev Genes Evol 214: 628–634.
Reed RD, Nagy L (2005). Evolutionary redeployment of a biosynthetic module: expression of eye pigment genes vermilion, cinnabar, and white in butterfly wing development. Evol Dev 7: 301–311.
Reed RD, Serfas MS (2004). Butterfly wing pattern evolution is associated with changes in a Notch/Distal-less temporal pattern formation process. Curr Biol 14: 1159–1166.
Richardson MK, Brakefield PM (2003). Hotspots for evolution. Nature 424: 894–895.
Sheppard PM, Turner JRG, Brown KS, Benson WW, Singer MC (1985). Genetics and the evolution of Muellerian mimicry in Heliconius butterflies. Philos Trans R Soc London Ser B 308: 433–610.
Stern DL (1998). A role of Ultrabithorax in morphological differences between Drosophila species. Nature 396: 463–466.
Suomalainen E, Cook LM, Turner JRG (1973). Achiasmatic oogenesis in the heliconiine butterflies. Hereditas 74: 302–304.
Tobler A, Flanagan N, Jiggins CD, Heckel DG, McMillan WO (2005). First-generation linkage map of the warningly colored butterfly Heliconius erato. Heredity 94: 408–417.
Turner JRG (1977). Butterfly mimicry: the genetical evolution of an adaptation. Evol Biol 10: 163–206.
Turner JRG (1984). Darwin's coffin and Doctor Pangloss – do adaptationist models explain mimicry? In: Shorrocks B (ed) Evol Ecol. Blackwell Scientific: Oxford. pp 313–361.
Turner JRG (1988). The evolution of mimicry: a solution to the problem of punctuated equilibrium. Am Nat 131: S42–S66.
Westerbergh A, Doebley J (2002). Morphological traits defining species differences in wild relatives of maize are controlled by multiple quantitative trait loci. Evolution 56: 273–283.
Wilkins AS (2002). The Evolution of Developmental Pathways. Sinauer: Sunderland.
Acknowledgements
We thank J Mallet, P Brakefield, S Baxter, M Kronforst and anonymous reviewers for their comments on the manuscript. MJ was funded by an EMBO long-term fellowship, CJ by a Royal Society University Research fellowship and grants from BBSRC and NERC, and WOM by the NSF (IBN-0344705) and the National Evolutionary Synthesis Center.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Joron, M., Jiggins, C., Papanicolaou, A. et al. Heliconius wing patterns: an evo-devo model for understanding phenotypic diversity. Heredity 97, 157–167 (2006). https://doi.org/10.1038/sj.hdy.6800873
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800873
Keywords
This article is cited by
-
Integrating ecological niche modeling and rates of evolution to model geographic regions of mimetic color pattern selection
Evolutionary Ecology (2024)
-
Seasonal Patterns of Host Plant Use in an Assemblage of Heliconiini Butterflies (Lepidoptera: Nymphalidae) in a Neotropical forest
Neotropical Entomology (2021)
-
Trait differentiation and modular toxin expression in palm-pitvipers
BMC Genomics (2020)
-
Genome-wide introgression among distantly related Heliconius butterfly species
Genome Biology (2016)
-
Assessing genotype-phenotype associations in three dorsal colour morphs in the meadow spittlebug Philaenus spumarius (L.) (Hemiptera: Aphrophoridae) using genomic and transcriptomic resources
BMC Genetics (2016)
