
Abstract
Background:
Mounting evidence points to the efficacy of donor lymphocyte infusion (DLI) and immunisation with tumour-pulsed dendritic cells (DC) in generating graft vs leukaemia reactions after allogeneic bone marrow transplantation (BMT). We assessed the efficacy of DLI and DC in generating potent graft vs neuroblastoma tumour (GVT) reactions following allogeneic BMT.
Methods:
Mice bearing congenic (H2Ka) Neuro-2a tumours were grafted with allogeneic (H2Kb) T-cell-depleted bone marrow cells. Tumour-pulsed donor DC (DCNeuro2a) were inoculated (on day +7) in conjunction with donor (H2Kb) and haploidentical (H2Ka/b) lymphocytes.
Results:
Murine Neuro-2a cells elicit immune reactions as efficient as B lymphoma in major histocompatibility complex antigen-disparate mice. Lymphopenia induced by conditioning facilitates GVT, and transition to adaptive immunity is enhanced by simultaneous infusion of and DCNeuro2a and lymphocytes devoid of graft vs host (GVH) activity (H2Ka/b). In variance, the efficacy of DC-mediated immunomodulation was diminished by severe graft vs host disease (GVHD), showing mechanistic dissociation and antagonising potential to GVT.
Conclsions:
The GVHD is not a prerequisite to induce GVT reactivity after allogeneic BMT, but is rather detrimental to induction of anti-tumour immunity by DC-mediated immunomodulation. Simultaneous inoculation of tumour-pulsed donor DC and DLI synergise in stimulation of potent GVT reactions to the extent of eradication of established NB tumours.
Similar content being viewed by others


Main
Neuroblastoma (NB) is a low immunogenic solid tumour that expresses low levels of major histocompatibility complex (MHC) antigens and few known specific antigens, most of which are shared with other tissues of ectodermal origin (Cooper et al, 1990; Coze et al, 1995; Prigione et al, 2004; Wolfl et al, 2005). Considering the poor outcome of surgery and chemoradiotherapy in paediatric patients with high-risk NB, immune-based therapeutic approaches are being pursued (Verneris and Wagner, 2007; Navid et al, 2009). For example, dendritic cells (DCs) are crucial ingredients towards induction and propagation of tumour antigen-specific immune reactions (Banchereau and Palucka, 2005), endowed with the capacity to sensitise the immune system against NB tumours ex vivo (Valteau-Couanet et al, 2002) and in vivo (Geiger et al, 2000; Caruso et al, 2005). Bone marrow transplantation (BMT) evolves as a standard therapeutic procedure aiming to maximise the efficacy and intensity of anti-tumour immunity (Fish and Grupp, 2008; Ladenstein et al, 2008). Consequently, immunomodulatory approaches have adopted to the transplant setting, consisting either of vaccination with tumour-pulsed DC (Asavaroengchai et al, 2002) or donor lymphocyte infusion (DLI) under conditions of controlled graft vs host disease (GVHD, Naparstek et al, 1996; Slavin, 2001).
Autologous BMT for immunohaematopietic reconstitution after aggressive chemoradiotherapy have yielded modest results in the absence of additional immunomodulation (Yaniv et al, 1990; Kanold et al, 2000). The modest efficacy might be attributed to the absence of graft vs tumour (GVT) reactions (Slavin et al, 2000), which is more efficiently attained by allogeneic and haploidentical BMT (Demirer et al, 2008; Kanold et al, 2008). Although allogeneic BMT per se is endowed with the capacity to generate anti-tumour immune reactivity (Ash et al, 2009), significant reduction and eradication of even small-sized NB tumours requires substantial post-transplant immunomodulation (Anderson et al, 2000; Borrello et al, 2000; Teshima et al, 2001; Asavaroengchai et al, 2002). Accordingly, DCs have been efficiently implemented to foster immunity against haematological malignancies after allogeneic transplantation (Li and Waller, 2004; Reddy et al, 2005; Tatsuta et al, 2009). However, haematological malignancies can elicit anti-tumour immunity through selective expression of minor histocompatibility antigens (Riddell et al, 2002), which are largely absent in solid tumours (Ringdén et al, 2009). In addition to effective immunisation against solid tumours using tumour-pulsed DC (Geiger et al, 2000; Caruso et al, 2005), recent evidence points to the superior activity of DC after allogeneic BMT (Moyer et al, 2006). Successful ex vivo sensitisation and expansion of tumour-reactive lymphocytes attribute DC a stimulatory effect (Xia et al, 2006; Ghosh et al, 2009), which can be achieved also by DLI (Eto et al, 2008; Kamiryo et al, 2009; Yoshida et al, 2009). Accordingly, the interaction between DC and lymphocytes (Chakraverty et al, 2006) has been implemented in a number of ways, including immunomodulation of the donors (Asavaroengchai et al, 2004) and immunomodulation of the recipients (Li and Waller, 2004; Moyer et al, 2006), although the relative roles of host and donor DC in GVT and GVH reactions is under debate (Shlomchik et al, 1999; Matte et al, 2004; Xia et al, 2006; Chakraverty and Sykes, 2007).
These data indicate that cytotoxic lymphocytes are effectively propagated by tumour-pulsed DC, with effective anti-leukaemia effects attained by post-transplant administration of lymphocytes and DC. Following the demonstration that tumour-pulsed donor DC effectively suppress NB growth after allogeneic BMT (S Ash, I Yaniv, N Askenasy, J Stein, unpublished data), it is questioned whether their effect can be amplified by DLI to elicit potent GVT reactions against this solid tumour. In variance from previous studies aiming to decipher the mechanism of interaction between antigen-presenting cells and lymphocytes (Shlomchik et al, 1999; Li and Waller, 2004; Matte et al, 2004; Chakraverty et al, 2006; Xia et al, 2006), we adopted a clinically relevant experimental model of post-transplant immunomodulation of tumour bearing mice using donor cells. We found that murine NB is an immunogenic tumour submitted to immune surveillance after allogeneic BMT through GVT reactivity that is independent of GVHD. Post-transplant immunisation with tumour-pulsed antigen-presenting cell in conjunction with lymphocytes devoid of GVH activity fosters significantly GVT reactivity to the extent of eradication of established tumours.
Materials and methods
Animal model
Mice used in this study were A/J (H2Ka, CD45.2), C57BL/6 (H2Kb, CD45.2), B6.SJL-Ptprca Pepcb/BoyJ (H2Kb, CD45.1), inbred A:C57BL/6 F1 chimeras (H2Ka/b, CD45.2) and non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice purchased from Jackson Laboratories (Bar Harbor, ME, USA). Mice were housed in a barrier facility in accordance with the guidelines of the Institutional Animal Care and Use Committee.
Tumour cells
Neuro-2a cells murine NB (H2Ka) and A20 B-cell lymphoma (H2Kd) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured to maximum 12 passages as previously described (Ash et al, 2009). Subcutaneous tumours were induced by implantation of 106 Neuro-2a cells in 100 μl of phosphate-buffered saline (PBS). Tumour growth was measured with a caliper and the volume (mm3) was calculated according to: (width2 × length × 0.4).
Bone marrow cell preparation
Whole bone marrow cells (wBMCs) were harvested from the femurs and tibia of donors, and low-density cells were collected as previously described (Askenasy and Farkas, 2003). Immunomagnetic T-cell depletion was performed by incubating for 45 min at 4°C with saturating amounts of biotinylated anti-mouse monoclonal antibodies (mAbs) specific to CD4, CD5 and CD8 (hybridoma cell cultures, ATCC). The mAb-coated cells were washed twice with PBS (Beit Haemek, Israel) containing 2% FCS (Biological Industries, Beit Haemek, Israel) and were incubated with sheep-anti-rat IgG conjugated to M-450 magnetic beads at a ratio of four beads per cell (Dynal Inc., Lake Success, NY, USA). Conjugated cells were precipitated by exposure to a magnetic field. The efficiency of T-cell depletion was reassessed by flow cytometry using a cocktail of primary labelled mAb against the T-cell markers listed above.
Splenocyte preparation
Spleens were harvested from mice, minced, passed through 40-μm mesh and dispersed into single-cell suspensions in PBS (Kaminitz et al, 2009). Red blood cells were lysed with medium containing 0.83% ammonium chloride, 0.1% potassium bicarbonate and 0.03% disodium EDTA. After 4 min, the reaction was arrested with excess of ice-cold PBS. T cells were enriched by elution through a cotton wool column (preferential retention of B lymphocytes and myeloid cells by differential charge than the eluted T cells), or immunomagnetic depletion using hybridoma-derived antibodies against GR-1, Mac-1 and B220 (ATCC). This procedure routinely provides an enriched population of 70–80% T cells.
Dendritic cells
Mononuclear cells were harvested from bone marrow samples by gradient centrifugation over murine Lympholyte (Cedarlane, Burlington, Ontario, Canada) and cultured (106 cells per ml) in low-LPS RPMI 1640 (<10 pg ml–1) supplemented with 10% FBS, 1% L-glutamine, 1% sodium pyruvate, 1% α-MEM non-essential amino acids, 0.1% Hepes buffer and 1% Pen/Strep (Biological Industries; Gibco BRL, Grand Island, NY, USA; Sigma, St Louis, MO, USA). The medium was supplemented with 50 ng ml–1 mouse recombinant (mr) granulocyte macrophage colony-stimulating factor (mrGM-CSF) and 10 ng ml–1 interleukin-4 (mrIL-4) on intermittent days (PeproTech, Rocky Hill, NJ, USA). Naïve DCs were incubated for additional 24 h in DC medium without growth factors. Pulsing with tumour antigens was performed by co-incubation of DC with tumour lysate at an approximate DC/tumour cell ratio of 3 : 1 (Ash et al, 2009). Tumour lysate is collected from the soluble fraction of Neuro-2a cells detached by trypsin, after three cycles of freezing in liquid nitrogen (2 min) and thawing at 37°C (4 min) in PBS.
Conditioning and transplantation
Recipients were conditioned with total body irradiation at 700 rad using an X-ray irradiator (Rad Source 2000; Rad Source Technologies Inc., Alpharetta, GA, USA) at a rate of 106 rad min–1. After 6 h, cells were injected into the lateral tail vein in 200 μl PBS. Naïve and tumour-pulsed DCs were inoculated subcutaneously adjacent to the tumour on day +7 after transplantation, and splenocytes were adoptively transferred intravenously.
Flow cytometry
Blood was collected in heparinised serum vials in 200 μl M199, centrifuged over 1.5 ml lymphocyte separation media (Cedarlane), and red blood cells were lysed. Nucleated cells were incubated for 45 min at 4°C with phycoerythrin (PE)-anti-H2Kb (Caltag, Carlsbad, CA, USA) and fluorescein isothiocyanate (FITC)-anti-H2Kk mAb cross-reactive with H2Ka (eBioscience, San Diego, CA, USA). Minor antigen disparity was assed using CD45.1-PE and CD45.2-FITC antibodies (eBioscience). T cells were quantified using CD4-allophycocyanin and CD8-FITC antibodies (BD Pharmingen, San Diego, CA, USA). Measurements were performed with a Vantage SE flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Positive staining was determined on a log scale, normalised with control cells stained with isotype control mAb.
Histology
Skin and liver were collected from mice killed by CO2 asphyxiation, and were fixed in ice-cold PBS containing 1.5% fresh paraformaldehyde for 2 h at 0–4°C. Tissues were embedded in OCT (Sakura Finetek, Torrance, CA, USA), frozen in isopentane suspended in liquid nitrogen, sectioned (3–6 μm) with a Cryotome (Termo Shandon, Cheshire, UK) and stained with haematoxylin and eosin.
GVH disease
Graft vs host disease was assessed using a semiquantitative clinical scale including weight loss, posture (hyperkeratosis of the foot pads impairs movement), activity, diffuse erythema (particularly of the ear) or dermatitis, and diarrhoea. GVHD was validated by histology in haematoxylin and eosin sections of the ear and liver according to: 0 – no infiltration, 1 – scarce infiltrates, 2 – patchy infiltration and 3 – diffuse infiltration with deterioration of tissue structure. The modified score for the liver (Fukui et al, 2007) is based on initial histological definition of GVHD pathology (Blazar et al, 1993; Cooke et al, 1998).
Proliferation assays
Splenocytes were layered in plastic dishes for 45 min and non-adherent cells were labelled with 2.5 μ M 5-(and-6-)-carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes, Eugene, OR, USA). Triplicate cultures were harvested after 3–5 days, evaluated for CFSE dilution by flow cytometry and data were quantified using the ModFit software (Verity Software House, Topsham, ME, USA). In some cases the cells were exposed to mitogenic stimulation with 5 μ M concanavalin A (Biological Industries).
Cytotoxic assays
Effector splenocytes harvested from naïve mice and chimeras were lysed and passed through wool mesh to enrich for T lymphocytes (∼70%). These cells were incubated with 5 × 105 Neuro-2a target cells for 7 h at 37°C in 150 μl at 1 : 10–1 : 100 target/effector ratios. Cytolysis was quantified by lactate dehydrogenase (eBioscience) release and normalised for background values.
Statistical analysis
Data are presented as means±s.d. for each experimental protocol. Results in each experimental group were evaluated for reproducibility by linear regression of duplicate measurements. Differences between the experimental protocols were estimated with a post hoc Scheffe t-test and significance was considered at P<0.05.
Results
Immunogenicity of Neuro-2a cells
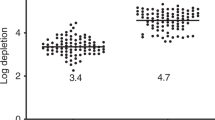
The Neuro-2a cell line is considered to be particularly low immunogenic, as a general feature of human NB. To determine the relative efficacy of tumour antigen presentation by Neuro-2a cells, cytolytic responses were compared with the murine A20 lymphoma cell line. B6 mice (H2Kb) were immunised twice at 3-day interval, and lymphocytes were co-incubated with target cells (Figure 1A). Neuro-2a cells (H2Ka) were gradually killed at decreasing target/effector ratios and were comparable to lysis of A20 (H2Kd, Figure 1B), with significant difference only at the lowest target/effector ratio (P<0.05). Therefore, the Neuro-2a cell line elicits effective immune responses comparable to an immunogenic cell line (Asavaroengchai et al, 2002, 2004).

Neuro-2a cells elicit immune reactions. (A) C57/BL mice (H2Kb) were immunised with two intravenous injections of 3 × 106 Neuro-2a (H2Ka) or A20 cells (H2Kd) at 3-day intervals and cytolytic responses were evaluated after 3 days. (B) Target Neuro-2a (n=6) and A20 cells (n=5) were exposed to lymphocytes from respectively immunised mice at various target/effector ratios to determine lysis according to lactate dehydrogenase (LDH) release. Data represent means±s.d.
Lymphopenia is favourable to the induction of GVT reactivity
Tumour growth in congenic mice is limited as compared with immunocompromised mice, suggesting that Neuro-2a is submitted to immune surveillance in immunocompetent mice (Ash et al, 2009). Furthermore, transition to adaptive immunity by either syngeneic or allogeneic BMT suppressed tumour growth, suggesting that a state of immune activation is beneficial to GVH reactivity. To evaluate whether lymphopenia actually promotes GVT reactivity (Borrello et al, 2000; Hu et al, 2002), tumour growth in reconstituted NOD/SCID mice was compared with allogeneic transplants (Figure 2A): mice were implanted with subcutaneous tumours and after 5 days were conditioned and grafted with 5 × 106 allogeneic BMC. NOD/SCID mice were conditioned with busulfan, which is significantly less immunosuppressive as compared with total body irradiation (Askenasy and Farkas, 2003). Tumour growth was significantly suppressed by transplantation of allogeneic whole BMC from H2Ka and K2Kb donors into NOD/SCID mice (P<0.01, Figure 2B), consistent with avid repopulation of the lymphopenic organs. Notably, residual host immune cells are detected in lymphoid organs of allografted immunocompetent mice after myeloablative irradiation (Kaminitz et al, 2009), whereas in NOD/SCID mice the lymphoid organs were full donor (not shown). Therefore, both intrinsic lymphopenia in NOD/SCID mice and radiation-induced lymphodepletion facilitate GVT reactivity.

Tumour growth in congenic and immunocompromised mice. (A) Mice were implanted with subcutaneous Neuro-2a cells and after 5 days were conditioned and grafted with 5 × 106 whole bone marrow cells (BMCs). Immunocompetent mice were irradiated (700 rad) and immunocompromised non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice were conditioned with busulfan (2 × 25 μg g–1). (B) Tumour growth in allografted mice congenic to the tumour (H2Ka, n=19) and NOD/SCID mice (H2Kg7) grafted with whole BMCs from B6 (H2Kb, n=9) and A (H2Ka, n=7) donors.
Relationship between GVT and GVHD
Enhanced GVT reactivity under transplant-associated lymphopenia questions whether immune reconstitution by DLI further augments anti-tumour immunity. We have recently observed that early administration of donor DC reduces tumour growth (S Ash, I Yaniv, N Askenasy, J Stein, unpublished data), whereas infusion of F1 splenocytes alone neither affect Neuro-2a growth in vivo nor induce GVHD as they are anergic to the host (Ash et al, 2009). The fast growth rates of Neuro-2a cells and the relatively fast tempo of immune reconstitution in mice impose early post-transplant immunomodulation in this experimental model. Mice bearing subcutaneous tumours were grafted with allogeneic (H2Kb → H2Ka) T-cell-depleted BMCs and on day +7 were inoculated with 106 donor DCs (subcutaneous) and 2 × 107 donor (H2Kb) and F1 (H2Ka/b) splenocytes (intravenous, Figure 3A). Whereas co-administration of donor lymphocytes resulted in higher tumour sizes as compared with DC alone (P<0.05, Figure 3B), F1 lymphocytes suppressed tumour growth (P<0.01 vs DC alone), resulting in remarkable superior efficacy of F1-DLI (P<0.005 vs allogeneic DLI). Furthermore, early allogeneic DLI induced high-grade GVHD (P<0.001, Figure 3C) accompanied by weight loss (P<0.001, Figure 3D), whereas F1 splenocytes were devoid of GVHD activity. These data dissociate the mechanisms of GVT and GVHD, emphasising that GVHD rather impairs the efficacy of GVT reactions.

Adoptive transfer of dendritic cells (DCs) and lymphocytes in allografted mice bearing tumours. (A) Experimental design of 5 × 106 allogeneic (H2Kb → H2Ka) T-cell-depleted bone marrow cell (TCD-BMC) transplantation into irradiated (700 rad) mice implanted with subcutaneous tumours 5 days earlier. On day +7 the mice were inoculated subcutaneously with tumour-pulsed DC (DCNeuro2a) in conjunction with intravenous infusion of 2 × 107 donor (H2Kb) or F1 (H2Ka/b) splenocytes (spln.). (B) Tumour growth in allografted mice (n=19), recipients of DCNeuro2a alone (n=17) and in conjunction with allogeneic (n=6) or F1 splenocytes (n=11). (C) Histological scoring of inflammatory infiltrates in ear wedge biopsies and livers of recipients of donor and F1 splenocytes. (D) Percent changes in body weight of recipients of allogeneic and F1 splenocytes. (E) Proliferation index of splenocytes upon mitogenic activation with concanavalin A (ConA) in vitro and rechallenge with naïve (DCNaive) and tumour-pulsed DC (DCNeuro2a): allografted mice (allogeneic bone marrow transplantation (allo-BMT)), recipients of F1 splenocytes alone (F1 spln.), recipients of tumour-pulsed DC (DCNeuro2a) alone and in conjunction with F1 splenocytes (n=6 in each group).
Donor DC present antigens in the context of compatible MHC to both allogeneic (H2Kb) and haploidentical splenocytes (H2Ka/b). The stimulatory effect of DC was evident from marked proliferative responses of lymphocytes to mitogenic stimulation at the experimental end point (4 weeks after BMT). Inoculation of tumour-pulsed DC alone enhanced the cycling rates of donor lymphocytes derived from the bone marrow of the chimeras, with a less accentuated effect after simultaneous administration of DC and F1 lymphocytes (P<0.05, Figure 3E). Importantly, in vitro rechallenge with naïve and tumour-pulsed DC generated strong proliferation (P<0.005 vs lymphocytes alone), emphasising effective DC-mediated immune activation in vivo as a basis for tumour growth suppression.
Consequences of DLI and DC administration
Given that the combination of F1 lymphocytes and DC suppressed tumour growth, we reasoned that fostering immune reconstitution by adoptive transfer of lymphocytes at the time of transplantation might provide additional benefit (Figure 4A). Sequential administration of F1 lymphocytes (day 0) and donor DC (day +7) conferred no significant inhibition of tumour growth (Figure 4B). Consistent with simultaneous administration of lymphocytes and DC (Figure 3E), proliferative responses were decreased by F1 lymphocyte infusion at the time of transplantation (P<0.05 vs DC alone, Figure 4C), and responsiveness was markedly increased by in vitro rechallenge with DC (P<0.005). Thus, DC administration has a stimulatory effect over lymphoid reconstitution; however, adoptive transfer of F1 lymphocytes in conjunction with BMCs does not suppress tumour growth.

Impact of adoptive transfer of F1 lymphocytes in conjunction with allogeneic bone marrow transplantation (allo-BMT). (A) Experimental setting of 2 × 107 F1 splenocyte (spln.) infusion concomitant with transplantation of 5 × 106 T-cell-depleted bone marrow cells (TCD-BMCs) (day 0) and subcutaneous administration of 106 tumour-pulsed dendritic cells (DCs) (DCNeuro2a) on day +7. (B) Tumour growth in recipients of F1 splenocytes (n=20), DCNeuro2a (n=17) and their joint administration (n=18). (C) Proliferation index of splenocytes upon mitogenic activation with concanavalin A (ConA) in vitro and rechallenge with naïve (DCNaive) and DCNeuro2a (n=5–6 in each group).
Reduced proliferation of splenocytes in recipients of F1 lymphocytes might be attributed either to the timing of adoptive transfer or variable interactions with the DC. These possibilities were analysed by monitoring the immune profiles of the reconstituted mice. Quantitatively, spleen cellularity at the experimental end point of 4 weeks was reduced by F1 lymphocyte infusion (day 0) and was apparently unaffected by F1 lymphocyte infusion on day +7 (Figure 5A). Qualitatively, minor differences in splenic T-cell subsets were observed under the different treatment protocols (Figure 5B), with a trend of reduced T-cell fraction in mice infused with DCNeuro2a. As F1 lymphocytes are not detected in the lymphoid organs of recipients at 4 weeks after transplantation (Ash et al, 2009), transient engraftment might modulate lymphoid reconstitution by cells derived from the bone marrow. Administration of F1 splenocytes on day 0 resulted in transient rise in spleen cellularity that subsided during the third week, whereas infusion of F1 splenocytes on day +7 caused a more sustained rise in spleen cellularity (Figure 5C). These differences correlate with the slower growth rates of tumours attained by F1 lymphocyte infusion on day +7 as compared with day 0, showing that the kinetics of lymphoid reconstitution are important to maximise the maturing effect of DC.

Kinetics of spleen reconstitution after bone marrow transplantation (BMT) and immunomodulation. (A) Percent recovery of spleen cellularity at 4 weeks after allogeneic BMT (allo-BMT) (H2Kb → H2Ka), infusion of F1 splenocytes (spln.) and tumour-pulsed dendritic cells (DCs) normalised against naïve C57/BL (B6) donors (n=5–6 for each point). (B) Fractional distribution of CD4+ and CD8+ T cells in the spleens of allografted mice in the various experimental protocols. (C) Dynamics of splenic cellularity in allografted mice and with adoptive transfer of F1 splenocytes on days 0 and +7 as a function of time elapsing from transplantation.
Efficacy of post-transplant immunomodulation
The relative efficacy of the various protocols was determined by tumour size at the experimental end point and eradication of established tumours (>60 mm3). Although tumour-pulsed DC suppressed the growth of Neuro-2a cells under all conditions (P<0.01, Figure 6A), adoptive transfer of F1 splenocytes alone had no significant impact. The only additive effect was obtained by joint administration of F1 splenocytes and DCNeuro2a on day +7 (P<0.01 vs DC alone, P<0.001 vs F1 splenocytes alone). Consistently, the profile of complete regression of tumours exceeding 60 mm3 mirrored the variations in tumour size (Figure 6B), reinforcing the validity of growth measurements. Notably, the experimental conditions were designed to maximise differences between the various experimental conditions by generating relatively large tumours. Therefore, the important component towards induction of effective GVT constitutes of donor DC infusion, which elicits potent reactions able to cause complete regression of established tumours.

Dendritic cells (DCs) enhance GVT reactivity. (A) Tumour size on day 28 after transplantation of allogeneic T-cell-depleted bone marrow cells (TCD-BMCs), subcutaneous inoculation of tumour-pulsed DC on day +7 and intravenous adoptive transfer of F1 splenocytes (spln.). (B) Complete regression of established tumours (>60 mm3) expressed as percentage of the experimental groups (numbers are given for each experimental group).
Discussion
Neuroblastoma is a potential target of GVT reactivity attained by allogeneic BMT and subsequent immunomodulation. Murine NB expresses MHC class II antigens (Ash et al, 2009) and elicits immune responses as efficient as A20 lymphoma in MHC-disparate mice (Asavaroengchai et al, 2002). However, effective GVT reactivity is mediated by cytotoxic cells derived from the bone marrow that are anergic to host MHC, indicating that tumour associated antigens are the primary targets of this reaction. Recruitment of lymphocytes reactive against the tumour is important in view of the low level expression of MHC antigens in human NB (Wolfl et al, 2005).
Transplant procedures associated with lymphopenia are generally associated with increased susceptibility to development of GVT reactions (Borrello et al, 2000; Hu et al, 2002). Our current data are consistent with the permissive effect of syngeneic and allogeneic BMT to evolution of anti-tumour immunity (Wrzesinski and Restifo, 2005), and the anti-tumoural activity of syngeneic transplants preceding implantation of tumours (Ash et al, 2009). In this study, we further demonstrate that the strain congenic to the tumour is not immunodeficient, as reconstitution of NOD/SCID mice impairs tumour growth as efficient as C57BL/6 donors. Effective tumour suppression upon transition to adaptive immunity by BMT might be particularly significant to the treatment of NB. This tumour develops in the prenatal phase from ectodermal origin (Coze et al, 1995; Prigione et al, 2004; Wolfl et al, 2005), and is therefore actively shielded by suppressive elements of innate immunity. This suppression is removed not only by pre-transplant conditioning, which is common to syngeneic and allogeneic transplants, but by the emerging GVT reactivity that develops during immunohaematopoietic reconstitution. Radiochemotherapy also inflicts direct injury to the tumour, facilitating selective sensitisation of the recovering immune system against tumour antigens (Asavaroengchai et al, 2002; Cho et al, 2000).
Professional processing and presentation of tumour antigens by DC elicits anti-tumour reactivity and causes complete regression of established NB tumours. Antigen-specific responsiveness is demonstrated by vigorous proliferative responses of lymphocytes upon rechallenge with tumour-pulsed DC in vitro. These data corroborate previous reports of potent vaccination following syngeneic BMT (Borrello et al, 2000; Asavaroengchai et al, 2002) achieved by tumour cells engineered to express co-stimulatory molecules (Jing et al, 2007) and cytokines (Bausero et al, 1995), and overexpression of cytokines in DCs (Tatsuta et al, 2009). A remarkable feature of DC-mediated tumour suppression was their efficacy at very low levels of T cells, reaching one-third of the steady state conditions at the experimental end point. Consistent with participation of DC in maturation of the developing immune system, the quantitative deficit was compensated by pronounced responses to antigens characteristic of lymphopenic mice as compared with T-cell-sufficient mice (Cho et al, 2000; Asavaroengchai et al, 2002; Shklovskaya and Fazekas, 2006).
In contrast to our initial assumption that early reinforcement of lymphoid reconstitution at the time of transplantation will favour GVT reactivity, simultaneous administration of F1 lymphocytes and donor DC converged to suppress tumour growth. This differential behaviour reflects distinct mechanisms of action, given that F1 lymphocytes engraft transiently and are undetected at 4 weeks after transplantation (Ash et al, 2009). Infusion of F1 lymphocytes at the time of BMT impaired spleen repopulation, likely through competitive homoeostatic expansion with BM-derived lymphocytes in peripheral lymphoid organs (Teshima et al, 2001; Zöller, 2003). In variance, immune activation by donor DC was fostered by F1 lymphocytes, cells endowed with cytotoxic activity against Neuro-2a cells (S Ash, I Yaniv, N Askenasy, J Stein, unpublished data). Therefore, transient lymphoid support conferred by DLI is beneficial to the generation of anti-tumour immunity, but the optimal timing of administration is significant (Chakraverty et al, 2006). Lymphocyte infusion appears to be necessary in the context of syngeneic BMT (Jing et al, 2009), whereas, in allogeneic transplants, DLI can have both positive and detrimental consequences. Although we applied immunomodulation early after transplantation, the toxicity of DLI is generally reduced when administered at later times (Eto et al, 2008; Prigozhina et al, 2008). Unlike the contention of reduced DC capacity to stimulate mature lymphocytes as compared with cells developing from the bone marrow (Feuerer et al, 2001), our data demonstrate effective transient stimulation of mature F1 lymphocytes despite dominance of the bone marrow in immune reconstitution (Ash et al, 2009). A transient beneficial effect of DLI has been recently documented in a case report in which infusion of ex vivo expanded donor CD4+ T cells mediated reactivity against disseminated recurrent NB (Yoshida et al, 2009).
Effective GVT reactivity attained by transplantation of allogeneic T-cell-depleted BMC is dissociated from the detrimental GVH disease (Anderson et al, 2000; Teshima et al, 2001; Lang et al, 2006; Ash et al, 2009). In our hands donor DC did not inflict GVHD, although their potential to enhance anti-host reactivity of immature bone marrow-derived lymphocytes and mature haploidentical (F1) lymphocytes has been demonstrated (Li and Waller, 2004; Matte et al, 2004; Banchereau and Palucka, 2005). Both GVT and GVH reactions are also effectively triggered by host DC (Shlomchik et al, 1999) and in conjunction with DLI (Chakraverty et al, 2006; Xia et al, 2006). Our data demonstrate that the tumour-suppressive reaction induced by donor DC was impaired by ongoing GVHD, suggesting little mechanistic association between these reactions. The GVHD is mediated primarily by mature donor lymphocytes that target minor tissue antigens (Ringdén et al, 2009), with severe toxicity in haploidentical transplants because the effectors are not rejected. It is therefore possible to dissociate between GVHD and therapeutic GVT, which is effectively attained through tumour-specific attack by host MHC-tolerant lymphocytes. Haploidentical TCD-BMT might be of benefit as a maximal possible mismatch for stimulation of the developing immune cells against malignant tissue, a clinically feasible approach when mature GVH effectors are eliminated form the donor inoculum, which can be achieved also in delayed DLI (Eto et al, 2008; Prigozhina et al, 2008; Bohana-Kashtan et al, 2009). The efficacy of host and donor DC in promoting the GVT reaction without imposing GVHD (Li and Waller, 2004; Chakraverty et al, 2006; Xia et al, 2006; Ghosh et al, 2009) suggest that non-myeloablative transplants might be equally and even more effective in generating anti-tumour immunity (Slavin et al, 2000).
In summary, NB is a solid tumour effectively attacked by immune reactions, which can be reinforced by post-transplant immunomodulation. As BMT becomes a standard procedure in high-risk patients, allogeneic transplants have the significant advantage of generating potent GVT reactions. Anti-tumour reactions are promptly initiated by DC at relatively early stages after transplantation under conditions of partial reconstitution of the T-cell compartment without causing GVHD, and can be augmented by DLI. These synergistic measures are significant in the competition against an aggressive and fast growing tumour such as NB, underlining the importance of optimisation of the details of the transplant procedure.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Anderson LD Jr, Savary CA, Mullen CA (2000) Immunization of allogeneic bone marrow transplant recipients with tumor cell vaccines enhances graft-vs-tumor activity without exacerbating graft-vs-host disease. Blood 95: 2426–2433
Asavaroengchai W, Kotera Y, Koike N, Pilon-Thomas S, Mulé JJ (2004) Augmentation of antitumor immune responses after adoptive transfer of bone marrow derived from donors immunized with tumor lysate-pulsed dendritic cells. Biol Blood Marrow Transplant 10: 524–533
Asavaroengchai W, Kotera Y, Mule JJ (2002) Tumor lysate-pulsed dendritic cells can elicit an effective antitumor immune response during early lymphoid recovery. Proc Natl Acad Sci USA 99: 931–936
Ash S, Gigi V, Askenasy N, Fabian I, Stein J, Yaniv I (2009) Graft vs neuroblastoma reaction is efficiently elicited by allogeneic bone marrow transplantation through cytolytic activity in the absence of GVHD. Cancer Immunol Immunother 58: 2073–2084
Askenasy N, Farkas DL (2003) In vivo imaging studies of the effect of recipient conditioning, donor cell phenotype and antigen disparity on homing of haematopoietic cells to the bone marrow. Br J Haematol 120: 505–515
Banchereau J, Palucka AK (2005) Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol 5: 296–306
Bausero M, Panoskaltsis-Mortari A, Blazar BR, Katsanis E (1995) Effective immunization against murine neuroblastoma using transduced tumor cells secreting GM-CSF and interferon-γ. Cancer Gene Ther 2: 309–317
Blazar BR, Taylor PA, Snover DC, Sehgal SN, Vallera DA (1993) Murine recipients of fully mismatched donor marrow are protected from lethal graft-vs-host disease by the in vivo administration of rapamycin but develop an autoimmune-like syndrome. J Immunol 151: 5726–5741
Bohana-Kashtan O, Morisot S, Hildreth R, Brayton C, Levitsky HI, Civin CI (2009) Selective reduction of graft-vs-host disease-mediating human T cells by ex vivo treatment with soluble Fas ligand. J Immunol 183: 696–705
Borrello I, Sotomayor EM, Rattis FM, Cooke SK, Gu L, Levitsky HI (2000) Sustaining the graft-vs-tumor effect through posttransplant immunization with granulocyte-macrophage colony-stimulating factor (GM-CSF)-producing tumor vaccines. Blood 95: 3011–3019
Caruso DA, Orme LM, Amor GM, Neale AM, Radcliff FJ, Downie P, Tang ML, Ashley DM (2005) Results of a phase I study utilizing monocyte-derived dendritic cells pulsed with tumor RNA in children with stage 4 neuroblastoma. Cancer 103: 1280–1291
Chakraverty R, Eom HS, Sachs J, Buchli J, Cotter P, Hsu R, Zhao G, Sykes M (2006) Host MHC class II+ antigen-presenting cells and CD4 cells are required for CD8-mediated graft-vs-leukemia responses following delayed donor leukocyte infusions. Blood 108: 2106–2113
Chakraverty R, Sykes M (2007) The role of antigen-presenting cells in triggering graft-vs-host disease and graft-vs-leukemia. Blood 110: 9–17
Cho BK, Rao VP, Ge Q, Eisen HN, Chen J (2000) Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J Exp Med 192: 549–556
Cooke KR, Hill GR, Crawford JM, Bungard D, Brinson YS, Delmonte J Jr, Ferrara JL (1998) Tumor necrosis factor- alpha production to lipopolysaccharide stimulation by donor cells predicts the severity of experimental acute graft-vs-host disease. J Clin Invest 102: 1882–1891
Cooper MJ, Hutchins GM, Mennie RJ, Israel MA (1990) Beta 2-microglobulin expression in human embryonal neuroblastoma reflects its developmental regulation. Cancer Res 50: 3694–3700
Coze C, Aalto-Setala K, Brenner M, Chiang Y (1995) Characteristics and immunomodulatory properties of human neuroblastoma cells after retrovirus-mediated gene transfer of the cytokine genes IL-2 and IFN-gamma. Transgenics 1: 585–595
Demirer T, Barkholt L, Blaise D, Pedrazzoli P, Aglietta M, Carella AM, Bay JO, Arpaci F, Rosti G, Gurman G, Niederwieser D, Bregni M (2008) Transplantation of allogeneic hematopoietic stem cells: an emerging treatment modality for solid tumors. Nat Clin Pract Oncol 5: 256–267
Eto M, Kamiryo Y, Takeuchi A, Harano M, Tatsugami K, Harada M, Kiyoshima K, Hamaguchi M, Teshima T, Tsuneyoshi M, Yoshikai Y, Naito S (2008) Posttransplant administration of cyclophosphamide and donor lymphocyte infusion induces potent antitumor immunity to solid tumor. Clin Cancer Res 14: 2833–2840
Feuerer M, Beckhove P, Bai L, Solomayer EF, Bastert G, Diel IJ, Pedain C, Oberniedermayr M, Schirrmacher V, Umansky V (2001) Therapy of human tumors in NOD/SCID mice with patient-derived reactivated memory T cells from bone marrow. Nat Med 7: 452–458
Fish JD, Grupp SA (2008) Stem cell transplantation for neuroblastoma. Bone Marrow Transplant 41: 159–165
Fukui J, Inaba M, Ueda Y, Miyake T, Hosaka N, Kwon AH, Sakaguchi Y, Tsuda M, Omae M, Kamiyama Y, Ikehara S (2007) Prevention of graft-vs-host disease by intra-bone marrow injection of donor T cells. Stem Cells 25: 1595–1601
Geiger J, Hutchinson R, Hohenkirk L, Mckenna E, Chang A, Mule J (2000) Treatment of solid tumours in children with tumour-lysate-pulsed dendritic cells. Lancet 356: 1163–1165
Ghosh A, Koestner W, Hapke M, Schlaphoff V, Länger F, Baumann R, Koenecke C, Cornberg M, Welte K, Blazar BR, Sauer MG (2009) Donor T cells primed on leukemia lysate-pulsed recipient APCs mediate strong graft-vs-leukemia effects across MHC barriers in full chimeras. Blood 113: 4440–4448
Hu HM, Poehlein CH, Urba WJ, Fox BA (2002) Development of antitumor immune responses in reconstituted lymphopenic hosts. Cancer Res 62: 3914–3919
Jing W, Gershan JA, Johnson BD (2009) Depletion of CD4 T cells enhances immunotherapy for neuroblastoma after syngeneic HSCT but compromises development of antitumor immune memory. Blood 113: 4449–4457
Jing W, Orentas RJ, Johnson BD (2007) Induction of immunity to neuroblastoma early after syngeneic hematopoietic stem cell transplantation using a novel mouse tumor vaccine. Biol Blood Marrow Transplant 13: 277–292
Kaminitz A, Mizrahi K, Yaniv I, Farkas DL, Stein J, Askenasy N (2009) Low levels of allogeneic but not syngeneic hematopoietic chmerism reverse autoimmune insulitis in prediabetic NOD mice. J Autoimmun 33: 83–91
Kamiryo Y, Eto M, Yamada H, Yajima T, Harano M, Takeuchi A, Tatsugami K, Hamaguchi M, Naito S, Yoshikai Y (2009) Donor CD4 T cells are critical in allogeneic stem cell transplantation against murine solid tumor. Cancer Res 69: 5151–5158
Kanold J, Paillard C, Tchirkov A, Merlin E, Marabelle A, Lutz P, Rousseau R, Baldomero H, Deméocq F (2008) Allogeneic or haploidentical HSCT for refractory or relapsed solid tumors in children: toward a neuroblastoma model. Bone Marrow Transplant 42 (Suppl 2): S25–S30
Kanold J, Yakouben K, Tchirkov A, Carret AS, Vannier JP, LeGall E, Bordigoni P, Deméocq F (2000) Long-term results of CD34(+) cell transplantation in children with neuroblastoma. Med Pediatr Oncol 35: 1–7
Ladenstein R, Pötschger U, Hartman O, Pearson AD, Klingebiel T, Castel V, Yaniv I, Demirer T, Dini G (2008) 28 years of high-dose therapy and SCT for neuroblastoma in Europe: lessons from more than 4000 procedures. Bone Marrow Transplant 41 (Suppl 2): S118–S127
Lang P, Pfeiffer M, Müller I, Schumm M, Ebinger M, Koscielniak E, Feuchtinger T, Föll J, Martin D, Handgretinger R (2006) Haploidentical stem cell transplantation in patients with pediatric solid tumors: preliminary results of a pilot study and analysis of graft vs tumor effects. Klin Padiatr 218: 321–326
Li JM, Waller EK (2004) Donor antigen-presenting cells regulate T-cell expansion and antitumor activity after allogeneic bone marrow transplantation. Biol Blood Marrow Transplant 10: 540–551
Matte CC, Liu J, Cormier J, Anderson BE, Athanasiadis I, Jain D, McNiff J, Shlomchik WD (2004) Donor APCs are required for maximal GVHD but not for GVL. Nat Med 10: 987–992
Moyer JS, Maine G, Mulé JJ (2006) Early vaccination with tumor-lysate-pulsed dendritic cells after allogeneic bone marrow transplantation has antitumor effects. Biol Blood Marrow Transplant 12: 1010–1019
Naparstek E, Nagler A, Or R, Kapelushnik J, Slavin S (1996) Allogeneic cell-mediated immunotherapy using donor lymphocytes for prevention of relapse in patients treated with allogeneic bone marrow transplantation for hematological malignancies. Clin Transpl 10: 281–290
Navid F, Armstrong M, Barfield RC (2009) Immune therapies for neuroblastoma. Cancer Biol Ther 8: 874–882
Prigione I, Corrias MV, Airoldi I, Raffaghello L, Morandi F, Bocca P, Cocco C, Ferrone S, Pistoia V (2004) Immunogenicity of human neuroblastoma. Ann NY Acad Sci 1028: 69–80
Prigozhina TB, Elkin G, Khitrin S, Slavin S (2008) Prevention of acute graft-vs-host disease by a single low-dose cyclophosphamide injection following allogeneic bone marrow transplantation. Exp Hematol 36: 1750–1759
Reddy P, Maeda Y, Liu C, Krijanovski OI, Korngold R, Ferrara JL (2005) A crucial role for antigen-presenting cells and alloantigen expression in graft-vs-leukemia responses. Nat Med 11: 1244–1249
Riddell SR, Murata M, Bryant S, Warren EH (2002) Minor histocompatibility antigens – targets of graft vs leukemia responses. Int J Hematol 76 (Suppl 2): 155–161
Ringdén O, Karlsson H, Olsson R, Omazic B, Uhlin M (2009) The allogeneic graft-vs-cancer effect. Br J Haematol 147: 614–633
Shklovskaya E, Fazekas de St GB (2006) Severely impaired clonal deletion of CD4+ T cells in low-dose irradiated mice: role of T cell antigen receptor and IL-7 receptor signals. J Immunol 177: 8320–8330
Shlomchik WD, Couzens MS, Tang CB, McNiff J, Robert ME, Liu J, Shlomchik MJ, Emerson SG (1999) Prevention of graft vs host disease by inactivation of host antigen-presenting cells. Science 285: 412–415
Slavin S (2001) Immunotherapy of cancer with alloreactive lymphocytes. Lancet Oncol 2: 491–498
Slavin S, Or R, Prighozina T, Gurevitch O, Aker M, Panighari S, Shapira M, Nagler A (2000) Immunotherapy of hematologic malignancies and metastatic solid tumors in experimental animals and man. Bone Marrow Transplant 25 (Suppl 2): S54–S57
Tatsuta K, Tanaka S, Tajiri T, Shibata S, Komaru A, Ueda Y, Inoue M, Hasegawa M, Suita S, Sueishi K, Taguchi T, Yonemitsu Y (2009) Complete elimination of established neuroblastoma by synergistic action of gamma-irradiation and DCs treated with rSeV expressing interferon-beta gene. Gene Ther 16: 240–251
Teshima T, Mach N, Hill GR, Pan L, Gillessen S, Dranoff G, Ferrara JL (2001) Tumor cell vaccine elicits potent antitumor immunity after allogeneic T-cell-depleted bone marrow transplantation. Cancer Res 61: 162–171
Valteau-Couanet D, Leboulaire C, Maincent K, Tournier M, Hartmann O, Benard J, Beaujean F, Boccaccio C, Zitvogel L, Angevin E (2002) Dendritic cells for NK/LAK activation: rationale for multicellular immunotherapy in neuroblastoma patients. Blood 100: 2554–2561
Verneris MR, Wagner JE (2007) Recent developments in cell-based immune therapy for neuroblastoma. J Neuroimmune Pharmacol 2: 134–139
Wolfl M, Jungbluth AA, Garrido F, Cabrera T, Meyen-Southard S, Spitz R, Ernestus K, Berthold F (2005) Expression of MHC class I, MHC class II, and cancer germline antigens in neuroblastoma. Cancer Immunol Immunother 54: 400–406
Wrzesinski C, Restifo NP (2005) Less is more: lymphodepletion followed by hematopoietic stem cell transplant augments adoptive T-cell-based anti-tumor immunotherapy. Curr Opin Immunol 17: 195–201
Xia G, Truitt RL, Johnson BD (2006) Graft-vs-leukemia and graft-vs-host reactions after donor lymphocyte infusion are initiated by host-type antigen-presenting cells and regulated by regulatory T cells in early and long-term chimeras. Biol Blood Marrow Transplant 12: 397–407
Yaniv I, Bouffet E, Irle C, Negrier S, Biron P, Favrot M, Philip I, Brunat-Mentigny M, Philip T (1990) Autologous bone marrow transplantation in pediatric solid tumors. Pediatr Hematol Oncol 7: 35–46
Yoshida H, Kusuki S, Hashii Y, Ohta H, Morio T, Ozono K (2009) Ex vivo-expanded donor CD4(+) lymphocyte infusion against relapsing neuroblastoma: a transient graft-vs-tumor effect. Pediatr Blood Cancer 52: 895–897
Zöller M (2003) Tumor vaccination after allogeneic bone marrow cell reconstitution of the nonmyeloablatively conditioned tumor-bearing murine host. J Immunol 171: 6941–6953
Acknowledgements
This work was supported by grants from the Israel Cancer Research Fund, Israel Ministry of Health, Israel Cancer Research Association and the Frankel Trust for Experimental Bone Marrow Transplantation. The excellent technical assistance of Mrs Ela Zuzovsky and Mrs Ana Zemlianski is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Ash, S., Stein, J., Askenasy, N. et al. Immunomodulation with dendritic cells and donor lymphocyte infusion converge to induce graft vs neuroblastoma reactions without GVHD after allogeneic bone marrow transplantation. Br J Cancer 103, 1597–1605 (2010). https://doi.org/10.1038/sj.bjc.6605924
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605924
Keywords
This article is cited by
-
Early immunisation with dendritic cells after allogeneic bone marrow transplantation elicits graft vs tumour reactivity
British Journal of Cancer (2013)
-
Recipient leukocyte infusion enhances the local and systemic graft-versus-neuroblastoma effect of allogeneic bone marrow transplantation in mice
Cancer Immunology, Immunotherapy (2013)
-
Allogeneic hematopoietic cell transplantation for neuroblastoma: the CIBMTR experience
Bone Marrow Transplantation (2013)
