
Abstract
Lomeguatrib, an O6-methylguanine-DNA methyltransferase inactivator, was evaluated in an extended dosing regimen with temozolomide, designed according to pharmacodynamic data from previous studies. Patients with unresectable stage 3 or 4 cutaneous or unknown primary melanoma metastases were treated with lomeguatrib 40 mg, b.i.d. for 10 or 14 days and temozolomide 75–100 mg m−2 on days 1–5. Drugs were administered orally with cycles repeated every 28 days, for up to six cycles. A total of 32 patients were recruited to the study. Lomeguatrib for 10 days with temozolomide 75 mg m−2 was established as the optimal extended lomeguatrib dosing schedule, with haematological toxicity being dose limiting. There were two partial responses to treatment giving an overall response rate of 6.25%. Extending lomeguatrib administration beyond that of temozolomide requires a reduced dose of the latter agent. Only limited clinical activity was seen, suggesting no advantage for this regimen over conventional temozolomide administration in the treatment of melanoma.
Similar content being viewed by others


Main
Treatment for metastatic melanoma is unsatisfactory: the standard of care, dacarbazine, offers modest response rates and has never been shown to improve patient survival compared with purely supportive care. Despite these shortcomings and over 30 years of randomised phase 3 trials no other agent, or combination of agents, has supplanted dacarbazine (Atkins, 1997; Balch et al, 1997).
Much effort has been devoted to understanding the intrinsic and acquired resistance of melanoma to chemotherapy. The most commonly applied treatments, temozolomide and dacarbazine, are methylating agents that share the active moiety methyltriazineimidazolecarboxamide. Their cytotoxicity is mediated principally through methylation of DNA at the O6 position of guanine (Ockey et al, 1986; Margison and O'Connor, 1990; Kaina et al, 1997). Repair of the lesion by O6-methylguanine-DNA methyltransferase (MGMT) and/or tolerance based on deficiencies in mismatch repair or the engagement of apoptosis have been identified as important aspects of resistance to dacarbazine and temozolomide (Branch et al, 1993; Friedman et al, 1998). New molecules and experimental approaches have followed but, as yet, none has proved superior to single agent therapy.
Tumour cells frequently express high levels of MGMT, and pre-clinical studies identify the protein as a key determinant of cell survival (Chen et al, 1992; Kaina et al, 1997). Inactivation of MGMT before dosing with an O6-alkylating agent considerably enhances the anti-tumour activity of the latter drug in vitro and in animal tumour models (Dolan et al, 1990; Karran and Bignami, 1994; Friedman et al, 2002). We have previously reported the development of lomeguatrib, a small molecule inactivator of MGMT, and its use in combination with temozolomide. In a phase 1 trial we established lomeguatrib 40 mg per day and temozolomide 125 mg m−2 per day p.o. for 5 consecutive days as well tolerated (Ranson et al, 2006). Total depletion of MGMT occurred in three of five subcutaneous melanoma metastases analysed 4 h after the first dose of lomeguatrib, and >96% depletion was observed in the other two.
A randomised phase 2 study failed to demonstrate any improved efficacy for lomeguatrib/temozolomide, nor any responses in patients treated with the combination after progression on temozolomide alone (Ranson et al, 2007). Melanoma biopsies taken in the 24–72 h after patients finished their first cycle of lomeguatrib and temozolomide showed residual MGMT activity. The daily dose of lomeguatrib was increased to 80 mg, which remained well tolerated. It transpired that there was rapid recovery of MGMT levels once dosing with lomeguatrib was completed, allowing the repair of O6-meG before the two rounds of DNA replication required for cytotoxicity could occur. The pharmacodynamic data suggested that the inactivator should be administered for several days after temozolomide treatment, and the current study was devised to determine the tolerability of the chemotherapy regimen when given with 10 or 14 days of lomeguatrib. The MGMT inactivator was given at 80 mg per day (40 mg, b.i.d.) as this had been well tolerated with temozolomide in the randomised phase 2 trial. In addition, tumour response rates, pharmacodynamics (reported in the accompanying paper), time to progression and survival were investigated.
Patients and methods
Inclusion and exclusion criteria
Patients with histologically proven cutaneous melanoma or unknown primary melanoma with metastases were eligible for the study, provided that they had not previously received systemic chemotherapy for melanoma. Other requirements included stage III or IV measurable disease, age >18 years, Eastern Cooperative Oncology Group performance status of 0 or 1, life expectancy >12 weeks, adequate bone marrow and biochemical function (haemoglobin >10 g per100 ml, white blood cells >3 × 109 per litre, absolute neutrophil count >1.5 × 109 per litre, platelets>100 000 per μl), creatinine ⩽1.25 upper limit of normal (ULN), bilirubin ⩽1.25 ULN, AST ⩽5 (metastases to liver) or 2 × ULN.
Patients were excluded if within 4 weeks of radio- or immunotherapy, pregnant or nursing, still recovering from surgery, suffering from significant comorbidity, had known brain metastases, had a history of seizures or were on anti-epileptic medication.
The study was conducted in accordance with the principles of the International Conference on Harmonisation of Good Clinical Practice guidelines and the Declaration of Helsinki Principles. The trial was approved by an independent ethics committee according to national and local requirements at each trial centre. All patients gave informed, written consent.
Study design and statistical considerations
The trial was a multi-centre, dose escalation study in which approximately 40 patients were to be enrolled. The aim of the study was to define the optimally tolerated dose of temozolomide given for 5 days with lomeguatrib given for 10 or 14 days. If one of three patients at a dose level developed dose-limiting toxicity, up to three additional patients were treated at that dose level. If one of the three additional patients developed a dose-limiting toxicity, dose escalation ceased and six patients were treated at the preceding dose level. This lower dose level was defined as the maximum tolerated dose unless ⩾2 of 6 patients developed dose-limiting toxicity. Dose-limiting toxicity was defined as any of the following events: grade 4 neutropaenia lasting >5 days or if associated with infection/fever, grade 4 thrombocytopaenia for >5 days, grade 3 or 4 non-haematological toxicity (excluding grade 3 nausea and vomiting in patients who had not received optimal treatment with anti-emetics) and drug-related death. Expansion at the optimally tolerated dose was mandated to allow a preliminary assessment of efficacy, toxicity, pharmacokinetic and pharmacodynamic end points. A second expansion, to 80 patients, was permitted should the effects of the optimally tolerated dose warrant further study. The median time to progression and median survival time were estimated using Kaplan–Meier survival curves. Patients who had not progressed by the end of the study or who withdrew before progression and patients who had not died were censored for the respective analyses.
Treatment
Lomeguatrib enteric-coated 10 mg capsules were obtained from Kudos Pharmaceuticals (Cambridge, UK), and temozolomide was purchased from Schering Plough Ltd (Welwyn Garden City, UK), as 5, 20, 100 and 250 mg capsules. Patients received lomeguatrib 40 mg, b.i.d. (p.o.) for 10 or 14 consecutive days every 4 weeks for up to six cycles. Temozolomide was administered on days 1–5 at 100 mg m−2 per day (p.o.) 2 h after lomeguatrib, with a planned escalation to 125 mg m−2 per day according to tolerability. Patients fasted for 1 and 2 h before and after temozolomide and lomeguatrib respectively.
A treatment delay of up to 2 weeks was allowed for resolution of drug-related toxicity. Dose reductions in temozolomide were mandated in the event of grade 4 haematological toxicity, grade 3 toxicity lasting 7 or more days or any grade 3 or 4 non-haematological toxicity. These were in decrements of 25 or 50 mg m−2 per day according to the type of toxicity encountered. Patients could be withdrawn for excessive toxicity, progressive disease, serious violation of the study drug protocol or withdrawal of consent.
Evaluation of response and toxicity
All eligible patients who received any part of the treatment were considered assessable for toxicity. Patients were assessed for adverse events at each attendance. Physical exam, performance status and vital signs were recorded at the beginning of each treatment cycle. Complete blood count was checked before treatment and on days 14, 21 and 28, with blood chemistry tested on days 1, 14 and 28. Patients who developed grade 4 bone marrow suppression were assessed every 1–2 days. Adverse events were graded according to the National Cancer Institute-Common Toxicity Criteria version 2.0. Tumour response was assessed every second cycle based on clinical and radiological findings in accordance with the RECIST criteria.
Pharmacodynamics
In a subset of patients peripheral blood mononuclear cell (PBMC) samples were obtained before treatment, 4 h after the first drug dose and at the end of treatment (day 10 or 14 depending on schedule) during cycle 1. Samples were assayed for MGMT activity, and levels of O6- and N7-methylguanine in DNA. Venous blood (5–10 ml) was collected into tubes containing 100 μl 0.5 M EDTA and stored on ice for maximum of 4 h before isolation of PBMC and analysis of MGMT (Watson and Margison, 2000).
Results
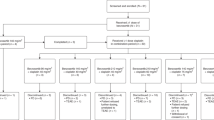
A total of 32 patients were enrolled in the study between August 2005 and May 2006, with characteristics as summarised in Table 1. All patients were evaluable for toxicity, but only 28 for efficacy. Four patients did not complete two cycles of treatment: three due to toxicity, one by choice.
Toxicity
Two patients died of causes other than melanoma while on treatment. In one instance the cause of death was unable to be ascertained by the study team, in the other the patient was admitted with a neutropaenic fever. Melanoma in the patient had progressed and the patient developed septic shock and multi-organ failure, classified as probably and possibly related to treatment respectively. A total of 86 cycles of treatment were administered, with 46 having to be delayed and seven individuals needing dose reductions. All the delays and dose adjustments were due to haematological toxicity.
In the first cohort, of 10 days lomeguatrib with temozolomide 100 mg m−2 per day, all three patients experienced significant myelotoxicity and required dose reductions and delays in treatment. Temozolomide 75 mg m−2 per day with 10 days lomeguatrib was better tolerated. A third cohort, with lomeguatrib dosing extended to 14 days was also recruited. Nine patients were treated in this last cohort, which was twice expanded to further assess feasibility beyond cycle 1 after proving just tolerable in the terms described in the protocol. Three patients experienced grade 4 neutropaenia and/or neutropaenic fever in cycle 1, and all but one required a delay in administering subsequent cycles. The second cohort was therefore expanded to allow a more accurate assessment of efficacy.
Although all patients had at least one treatment-emergent adverse event, therapy was well tolerated aside from the increased haematological toxicity noted in cohorts 1 and 3. All treatment-related serious adverse events were neutropaenic episodes associated with fever and/or infection. The most frequent non-haematological toxicities attributed to treatment are summarised in Table 2, and were in keeping with those described for temozolomide alone.
Efficacy
Of the 32 patients treated, 28 were evaluable for response. There were two partial responses to treatment, one each in cohorts 1 and 2, giving an overall response rate of 6.25%. A further 10 patients had stable disease, but in only 4 cases did this extend beyond the second assessment at the end of cycle 4. Median time to progression was 62 days, and median overall survival was 264 days. In view of the lack of efficacy the second expansion phase of the trial was not performed.
Pharmacodynamics
Peripheral blood mononuclear cell samples were obtained from 10 patients during the course of the study. These were analysed for levels of DNA damage, specifically O6- and N7-methylguanine, and for MGMT expression. Detailed results are presented in the accompanying paper.
Discussion
Prolonged inactivation of MGMT in this patient group failed to improve upon the activity expected from temozolomide alone. The response rate of 6.25% with combination therapy is disappointing, being approximately half that seen in the phase 3 comparison of temozolomide and dacarbazine and our earlier phase 2 trial of lomeguatrib and temozolomide – trials with similar entry criteria to the current study (Middleton et al, 2000; Ranson et al, 2007). Results were very similar to those achievable with temozolomide alone: median progression free and overall survival were 62 and 264 days, compared with 58 and 241 days in the phase 3 study.
From the point of view of toxicity, the patients fared as expected. The combination of lomeguatrib and temozolomide was well tolerated: although haematological toxicity was more frequent it was readily managed. Non-haematological side effects were very similar to those seen for the methylating agent alone, with nausea and vomiting, constipation, headache and fatigue predominant (Middleton et al, 2000).
The rationale for the current study came from the phase 2 trial involving lomeguatrib and temozolomide, where we observed recovery of tumour MGMT activity within 24 h of completing treatment (Ranson et al, 2007). In that trial the efficacy of lomeguatrib and temozolomide was no better than that of the cytotoxin alone. O6-methylguanine is toxic only through replication and may be repaired effectively at any point beforehand, so one possible explanation was that the recovery of tumour MGMT levels occurred before successive rounds of replication could cause cell death (Ollila et al, 1998).
As measured in PBMCs, extended dosing with lomeguatrib successfully depleted MGMT for the duration of treatment, and resulted in the persistence of DNA damage. There was an evident impact on bone marrow progenitor cells, because continued suppression of MGMT activity required a reduction in the dose of temozolomide given over 5 days due to haematological toxicity. Used alone a total of 1000 g m−2 temozolomide is deliverable, but with 5 days lomeguatrib only 625 mg m−2 is possible. Extending the lomeguatrib dosing to 10 days requires a further reduction to 375 mg m−2 temozolomide over the 5 days, and this dose is barely tolerable with 14 days of the inactivator (Middleton et al, 2000; Ranson et al, 2006, 2007).
Despite the effects of the MGMT inactivator on enhancing bone marrow toxicity, we failed to see a similar effect on melanomas, suggesting that tolerance of damage and/or failure of downstream elements required for cytotoxicity have greater importance in melanoma cells. It seems likely that other survival pathways prevent increased DNA damage from killing melanoma cells. For example, several defects in the apoptotic machinery have been reported in melanoma, such as upregulation of bcl-2, or epigenetic silencing of DAP kinase and Apaf-1. Another possible explanation for the difference between tumour and bone marrow is that tumour doubling times are such that the extension of MGMT inactivation to 10 or 14 days is still not sufficient to allow the two rounds of replication required for cell killing. Indeed in one series of patients studied for consideration of pulmonary metastasectomy, median tumour doubling time was 66.9 days (Ollila et al, 1998).
Our results, and those of others using O6-benzylguanine (Gajewski et al, 2005), hold out little hope for MGMT inactivation in the effective treatment of melanoma. Despite the wealth of pre-clinical data to support MGMT's role in determining the outcome of methylating agent treatment in melanoma, evidence for the protein's importance in the clinic is inconclusive (Middleton et al, 1998; Ma et al, 2002; Middleton and Margison, 2003). More might be achievable with better targeting of inactivation to tumour, but such agents are not yet available clinically. It also remains to be seen whether inactivation might offer a benefit in other tumour types, such as glioblastoma, where temozolomide is an active agent and the importance of MGMT in determining chemoresistance has been established (Hegi et al, 2005). In conclusion, administration of an extended schedule of lomeguatrib with temozolomide confers no advantage over conventional temozolomide dosing in melanoma.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Atkins MB (1997) The role of cytotoxic chemotherapeutic agents either alone or in combination with biological response modifiers,. In Molecular Diagnosis, Prevention & Therapy of Melanoma Kirkwood, Kirkwood JK (ed), pp 219–251. New York: Marcel Dekker
Balch CM, Reintgen DS, Kirkwood JM, Houghton A, Peters L, Ang KK (1997) Cutaneous melanoma, In Cancer: Principles and Practice of Oncology (5th edn), De Vita VT Jr, Hellman S, Rosenberg SA (eds), pp 1947–1994. Philadelphia, PA: Lippincott-Raven
Branch P, Aquilina G, Bignami M, Karran P (1993) Defective mismatch binding and a mutator phenotype in cells tolerant to DNA damage. Nature 362: 652–654
Chen J, Zhang Y, Wang C, Sun Y, Fujimoto J, Ikenaga M (1992) O6-methylguanine-DNA methyltransferase activity in human tumors. Carcinogenesis 13: 1503–1507
Dolan ME, Moschel RC, Pegg AE (1990) Depletion of mammalian O6-alklguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc Natl Acad Sci USA 87: 5368–5372
Friedman HS, Keir S, Pegg AE, Houghton PJ, Colvin OM, Moschel RC, Bigner DD, Dolan ME (2002) O6-benzylguanine-mediated enhancement of chemotherapy. Mol Cancer Ther 1: 943–948
Friedman HS, McLendon RE, Kerby T, Dugan M, Bigner SH, Henry AJ, Ashley DM, Krischer J, Lovell S, Rasheed K, Marchev F, Seman AJ, Cokgor I, Rich J, Stewart E, Colvin OM, Provenzale JM, Bigner DD, Haglund MM, Friedman AH, Modrich PL (1998) DNA mismatch repair and O6-alkylguanine-DNA alkyltransferase analysis and response to Temodal in newly diagnosed malignant glioma. J Clin Oncol 16: 3851–3857
Gajewski TF, Sosman J, Gerson SL, Liu L, Dolan E, Lin S, Vokes EE (2005) Phase II trial of the O6-alkylguanine DNA alkyltransferase inhibitor O6-benzylguanine and 1,3-bis(2-chloroethyl)-1-nitrosourea in advanced melanoma. Clin Cancer Res 11: 7861–7865
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352: 997–1003
Kaina B, Ziouta A, Ochs K, Coquerelle T (1997) Chromosomal instability, reproductive cell death and apoptosis induced by O6-methylguanine in Mex−, Mex+ and methylation-tolerant mismatch repair compromised cells: facts and models. Mutat Res 381: 227–241
Karran P, Bignami M (1994) DNA damage tolerance, mismatch repair and genome instability. Bioessays 16: 833–839
Ma S, Egyházi S, Martenhed G, Ringborg U, Hansson J (2002) Analysis of O(6)-methylguanine-DNA methyltransferase in melanoma tumours in patients treated with dacarbazine-based chemotherapy. Melanoma Res 12 (4): 335–342
Margison GP, O'Connor PJ (1990) Biological consequences of reactions with DNA: role of specific lesions, In Handbook of Experimental Pharmacology, Cooper CS, Grover P (eds), pp 547–571. Berlin: Springer-Verlag
Middleton MR, Grob JJ, Aaronson N, Fierlbeck G, Tilgen W, Seiter S, Gore M, Aamdal S, Cebon J, Coates A, Dreno B, Henz M, Schadendorf D, Kapp A, Weiss J, Fraass U, Statkevich P, Muller M, Thatcher N (2000) A randomized, phase III study of temozolomide (TMZ) versus dacarbazine (DTIC) in the treatment of patients with advanced, metastatic malignant melanoma. J Clin Oncol 18: 158–166
Middleton MR, Lunn JM, Morris C, Rustin G, Wedge SR, Brampton MH, Lind MJ, Lee SM, Newell DR, Bleehen NM, Newlands ES, Calvert AH, Margison GP, Thatcher N (1998) O6-methylguanine-DNA methyltransferase in pre-treatment tumour biopsies as a predictor of response to temozolomide in melanoma. Br J Cancer 78: 1199–1202
Middleton MR, Margison GP (2003) Enhancing chemotherapy by inactivation of a DNA repair pathway. Lancet Oncol 4: 37–44
Ockey CH, White GRM, Brennand J, Margison GP (1986) Chinese hamster cells harbouring the E. coli O6-alkylguanine alkyltransferase gene are less susceptible to SCE induction and chromosome damage by methylating agents. Carcinogenesis 7: 2077–2080
Ollila DW, Stern SL, Morton DL (1998) Tumor doubling time: a selection factor for pulmonary resection of metastatic melanoma. J Surg Oncol 69: 206–211
Ranson M, Hersey P, Thompson D, Beith J, McArthur GA, Haydon A, Davis ID, Kefford RF, Mortimer P, Harris PA, Baka S, Seebaran A, Sabharwal A, Watson AJ, Margison GP, Middleton MR (2007) A randomised trial of the combination of lomeguatrib and temozolomide and temozolomide alone in patients with advanced melanoma. J Clin Oncol 25: 2540–2545
Ranson M, Middleton MR, Bridgewater J, Lee SM, Dawson M, Jowle D, Halbert G, Waller S, McGrath H, Gumbrell L, McElhinney RS, Donnelly D, McMurry TB, Margison GP (2006) Lomeguatrib, a potent inhibitor of O6 alkylguanine DNA alkyltransferase: phase I safety, pharmacodynamic and phamacokinetic trial and evaluation in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res 12: 1577–1584
Watson AJ, Margison GP (2000) O6-alkylguanine-DNA alkyltransferase assay. Methods Mol Biol 152: 49–61
Acknowledgements
Part of this work was supported by a grant from the European Commission (LSHC-CT-2007-037665), and by the NIHR Biomedical Research Centre Programme.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Kefford, R., Thomas, N., Corrie, P. et al. A phase I study of extended dosing with lomeguatrib with temozolomide in patients with advanced melanoma. Br J Cancer 100, 1245–1249 (2009). https://doi.org/10.1038/sj.bjc.6605016
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605016
Keywords
This article is cited by
-
Unboxing the molecular modalities of mutagens in cancer
Environmental Science and Pollution Research (2022)
-
Temozolomide–perillyl alcohol conjugate downregulates O6-methylguanin DNA methltransferase via inducing ubiquitination-dependent proteolysis in non-small cell lung cancer
Cell Death & Disease (2018)
-
Chemotherapeutic resistance in anaplastic astrocytoma cell lines treated with a temozolomide–lomeguatrib combination
Molecular Biology Reports (2014)
-
Inhibition of DNA repair with MGMT pseudosubstrates: phase I study of lomeguatrib in combination with dacarbazine in patients with advanced melanoma and other solid tumours
British Journal of Cancer (2011)
-
DNA repair pathways and their implication in cancer treatment
Cancer and Metastasis Reviews (2010)
