
Abstract
Systemic chemotherapy for gastric cancer is often associated with treatment-related toxicity, which is particularly severe in patients with a poor performance status. In this paper, we describe the first study to evaluate S-1 monotherapy as an option for advanced gastric cancer patients who are not candidates for combination chemotherapy due to poor clinical condition. Fifty-two patients with Eastern Cooperative Oncology Group (ECOG) performance scale 2–3, whose general condition had made use of combination chemotherapy impossible, were enrolled. S-1 was administered to 30 patients as second- or third-line therapy. The initial dose of S-1 was 35 mg m−2, administered b.i.d for 14 days every 3 weeks. With a median follow-up period of 33 weeks, the median progression-free survival, and overall survival were 11 weeks (95% CI, 8–14) and 33 weeks (95% CI, 19–47), respectively. The overall 1-year survival rate was 29% by intent-to-treat analysis. The overall response rate was 12% (95% CI, 3–21), and the percentage of stable disease was 35%, resulting in the disease control rate of 47% (95% CI, 32–60). Significant drug-related toxicity included grade 3 diarrhoea (14%), anorexia (14%), fatigue (10%), neutropenia (10%), and leucopenia (6%). In conclusion, this study indicated the modest activity of S-1 in gastric cancer patients with poor performance status.
Similar content being viewed by others
Main
Performance status is a widely accepted parameter that predicts response to chemotherapy and survival, and it is associated with many factors such as age, previous chemotherapy, and comorbidity (Lavin et al, 1982; Janunger et al, 2001; Wilson et al, 2005). In advanced gastric cancer, patients are likely to suffer from poor general condition due to anorexia and weight loss, often as a consequence of peritoneal carcinomatosis. These patients have been excluded from clinical trials, and chemotherapeutic options for them are quite limited (Cunningham et al, 1987; Hsu et al, 1997).
Combination chemotherapy is a standard approach in advanced gastric cancer, with many combination regimens showing a response rate of 35–45% for first-line treatment. However, these regimens are inevitably accompanied by substantial toxicities, which reduce their value as a palliative treatment. This toxicity is particularly significant in patients whose performance status is compromised. S-1 is a fourth-generation oral fluoropyrimidine that was developed to mimic protracted continuous infusion of 5-fluorouracil (5-FU). In the phase II trials conducted in Japan, S-1 monotherapy demonstrated promising activity which was comparable to combination chemotherapy in advanced gastric cancer, and it has been one of the preferred agents for gastric cancer (Sakata et al, 1998; Sugimachi et al, 1999; Koizumi et al, 2000). Moreover, it has a safety profile, which is favourable compared with other oral fluoropyrimidines used in gastric cancer. In addition, single agent 5-FU has been an option for patients with poor general condition, and also for patients complicated with disseminated intravascular coagulation (Chao et al, 2000).
Based on these observations, S-1 may be a substitute for the conventional chemotherapy in patients with poor general condition. We conducted the first prospective study to evaluate the feasibility of S-1 as an option for gastric cancer patients who were not candidates for more intensive chemotherapy due to poor clinical condition.
Patients and methods
Patient eligibility
This study was designed as a single-institutional phase II trial. Patients with histologically proven metastatic and/or relapsed gastric adenocarcinoma were considered eligible for the study when they met all of the following criteria: aged ⩾18; Eastern Cooperative Oncology Group (ECOG) performance scale 2–3; assessable disease with or without measurable lesion; either chemotherapy-naïve or having completed chemotherapy due to disease progression within 3 months before entry; and adequate haematological, renal, and hepatic functions. The latter was defined as neutrophil ⩾1500 μl−1, platelet ⩾75 000 μl−1, serum creatinine ⩽1.5 mg dl−1, total bilirubin ⩽1.25 (or 1.5) × upper limit of normal (ULN), and serum transaminases ⩽2.5 (or 5.0) × ULN in the absence (or presence) of liver metastasis. Patients were excluded from the study if they had concurrent cancer within the past 3 years (excluding basal cell carcinoma of the skin or cervical carcinoma in situ), active metastasis to central nervous system, or uncontrolled significant comorbid conditions. After the protocol was approved by the institutional review board, written informed consent with ICH Guidelines was obtained from patients.
Treatment schedule
The starting dose of S-1 was 35 mg m−2 twice daily (b.i.d). S-1 was administered within 1 h after the morning and evening meals for 14 consecutive days, followed by a 7-day resting period. The S-1 dosage assigned to the patients was calculated based on body surface area, which was different from the Japanese dosing system. The planned dose intensity was 327 mg m−2 week−1. The schedule was repeated until the occurrence of disease progression, unacceptable toxicities, or patient's refusal. In the absence of evidence of disease progression, the patients were allowed to continue S-1 treatment to the maximum 12 cycles. A dose reduction of 10 mg m−2 a day was made if ⩾grade 3 haematological or non-haematological toxicity was shown in the previous cycle. Dose re-escalation was not allowed. Patients who required more than 4 weeks of rest for recovery from any toxicity other than alopecia, nausea, vomiting, or anaemia, or who required dose reduction of more than two steps (total 20 mg m−2 a day) were withdrawn from the study.
Evaluation of response and toxicity
Weight loss, comorbidity, and performance status at presentation were established by direct questioning of the patient during a preliminary assessment by a physician at the first attendance for S-1 treatment. Baseline evaluations of each patient included complete medical history with physical examination, complete blood count (CBC), serum chemistry, urine analysis, and electrocardiography. A radiological evaluation was completed within 3 weeks before treatment. Fibre-optic gastroduodenoscopy and positron-emission tomography were planned to examine complete response (CR) of all measurable lesions. During treatment, patients were evaluated by a weekly CBC. A physical examination including weight, performance status, and serum chemistry were performed before each subsequent cycle. Radiological studies were repeated every two cycles.
Treatment response was evaluated by spiral CT scan according to the guidelines of the Response Evaluation Criteria in Solid Tumors (RECIST) committee. The response was analysed according to an intent-to-treat (ITT) analysis. A measurable lesion was defined as 10 mm in the longest dimension. If a patient was documented as having a CR or a partial response (PR), the response was confirmed at least 4 weeks after the first evidence of response.
Progression-free survival (PFS) was defined as the time elapsed from the start of treatment until disease progression or death of any cause, and overall survival (OS) was defined as the start of treatment to death. Time-to-treatment-failure was defined as the time interval between treatment initiation and cessation due to any reason. All patients were evaluated for toxicity from the time of their first cycle. Toxicity was evaluated as a grade according to the NCI-CTC (version 2.0).
Biostatistics
The primary aim of this study was to test the hypothesis that 1-year survival would improve by what degree compared to historical controls. The study was designed to have an 80% power to show an improvement in 1-year survival rate to 30% from historical control of 15 with 5% type-I error, using two-sided testing, and assuming exponential overall survival times. According to Minimax phase II design, a sample size of 48 patients was required. Considering a 5% drop-out rate, 52 patients were needed for this trial. The secondary aims included response rate in patients according to RECIST criteria, safety, PFS, time-to-treatment-failure, and OS. Time-dependent variables were analysed using the Kaplan–Meier method and compared using the log–rank test. Multivariate analysis was performed using Cox's proportional hazard regression model. Exact 95% confidence interval (CI) was provided for proportions.
Results
Patient characteristics
A total of 52 patients were enrolled between August 2004 and July 2006. Baseline patient characteristics are provided in Table 1. All patients were evaluable for survival. The median age was 61 years, including 12 patients (23%) who were ⩾70 years old. Twenty-four patients (46%) had performance status of grade 3. Median value of weight loss from the diagnosis of gastric cancer to S-1 start was 7% (range 5–23%) and 21 patients (40%) suffered more than 10% weight loss. Twenty-six patients had comorbidity other than gastric cancer. More than half of the patients received prior first- or second-line palliative chemotherapy. Twenty-two patients (42%) had no history of palliative chemotherapy. Site of metastasis commonly included peritoneum (45%), abdominal lymph nodes (33%), and stomach (33%).
Table 2 summarises the prior chemotherapy which our patients received. Twenty-one patients (40%) received prior adjuvant chemotherapy after curative resection. The median time elapsed from documentation of disease progression (or relapse) of previous regimen to S-1 treatment was 36 days (range 14–73). The median number of cycles of previous chemotherapy was eight (range 2–24) per patient.
Treatment outcomes
The median dose administered per day was 100 mg (range 80–130). A total of 233 treatment cycles (median 2.5, range 1–12) were administered. Six patients (12%) completed the planned 12 cycles. Four patients were subjected to dose reduction by one step, and two patients were by two steps due to grade 3 adverse events. Total 31 cycles were delayed in 16 patients with the median delayed duration of 1 week (range 1–4). The reason of delay was as follows: non-haematological toxicity in 13 cycles; haematological toxicity in 10 cycles; and the patients' will in the remaining eight cycles. Finally, the median dose intensity of all patients was 308 mg m−2 week−1 (range 163–327), which the relative dose intensity was 94%. Eighteen patients (35%) were transferred to the other chemotherapy regimens after disease progression was documented.
Survival
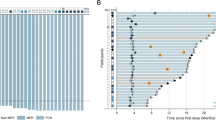
At the time of analysis, all the patients finished S-1 treatment. With the median follow-up period of 33 weeks (range 3–98), 46 patients showed disease progression, and all but eight patients (85%) expired. Median overall survival duration was 33 weeks (95% CI, 19–47), and the 1-year survival rate was 29%. Median PFS was 11 weeks (95% CI, 8–14) (Figure 1). Patients with measurable disease (n=30) demonstrated a median PFS of 10 weeks (95% CI, 2–17) while patients with assessable, but non-measurable disease (n=22) had a median PFS of 19 weeks (95% CI, 5–33) (P=0.12).

Survival analysis of all patients (N=52).
When the PFS was analysed with respect to prior chemotherapy, among patients who received S-1 as first-line treatment the median PFS was 18 weeks (95% CI, 7–23), whereas second- or third-line patients showed 8 weeks (95% CI, 1–15) (P=0.07). Median time-to-treatment-failure of all the patients was 9 weeks (95% CI, 14–104).
When we compared the PFS profile according to clinical parameters by multivariate analysis, patients' age (>65) (P=0.015), and previous exposure to chemotherapy (P=0.018) were the significant factors for poor PFS for measurable disease. For non-measurable disease, no factor was found to be significant for PFS. Severe weight loss (grade >2) (P=0.008), male sex (P=0.02), previous exposure to chemotherapy (P=0.012), and prior gastrectomy (P=0.03) were independent factors for poor overall survival.
Response
Response to therapy was assessable in all but one patient, who withdrew the consent after the first cycle (Table 3). The overall response rate was 12% (95% CI, 3–21), and the percentage of stable disease was 35%, resulting in the disease control rate of 47% (95% CI, 32–60). Of 30 patients with measurable disease, three patients achieved a PR, and one had a CR giving an overall response rate of 13% by ITT analysis. For 22 patients with non-measurable disease, two patients showed CR, and nine patients showed non-progressive disease. All but eight patients stopped S-1 treatment because of disease progression. Forty-four patients had PD involving progression of pre-existing lesions, while the remaining patients (15%) showed new lesions: peritoneal seeding (n=3), lung (n=2), brain (n=1), liver (n=1), and bile duct (n=1). When the response was analysed with respect to prior chemotherapy, among patients who received S-1 as first-line treatment there were five responses (23%), whereas 30 second- or third-line patients showed only one CR without a PR (3%) (P=0.07). When the response was also analysed according to performance status, in ECOG two patients there were three responses (11%), and also in ECOG three patients, there were three responses (13%).
Toxicity
Toxicity was assessable in all 52 patients (Table 4). There was one case of sudden death during treatment of an 81-year old man with a history of hypertension who received six courses of treatment. There was no evidence of disease progression or toxicity in this patient, but there occurred sudden cardiac death in the resting period of cycle 6. Common significant drug-related toxicity included grade 3 diarrhoea (14%), anorexia (14%), fatigue (10%), neutropenia (10%), and leucopenia (6%). Two patients developed febrile neutropenia (4%). Three patients suffered weight loss of more than 10% during S-1 treatment. Seven hospitalisations possibly related to treatment occurred in six patients (12%); gastrointestinal toxicity (n=6), and aspiration pneumonia accompanied with febrile neutropenia (n=1).
Discussion
To the best of our knowledge, this is the first prospective report on S-1 treatment in gastric cancer patients with poor performance status. Old age, poor performance status, comorbidity, and significant weight loss are adverse prognostic factors for gastric cancer (Andreyev et al, 1998; Trumper et al, 2006). Regimens of cisplatin, doxorubicin, or etoposide are associated with severe drug-related toxicity that could offset the potential clinical benefit. Recently developed chemotherapy regimens including taxanes, irinotecan, and oxaliplatin have better safety profiles compared to the older regimens. However, the benefit of these new regimens has not been evaluated in patients with poor performance status because such patients are traditionally excluded from clinical trials.
The definition of poor general condition differs among studies, but the first chemotherapeutic consideration for poor general condition was the ELF regimen (etoposide, 5-FU, leucovorin) (Wilke et al, 1990). This regimen showed favourable compliance for elderly patients or those with cardiac disease preventing doxorubicin treatment, but 20% of patients suffered from grade 3–4 neutropenia and 7% from grade 3 diarrhoea. High-dose 5-FU with leucovorin (HDFL) showed promising efficacy in patients with poor general condition of old age, performance status, comorbidity, malnutrition, and cytopenia (Hsu et al, 1997). However, this regimen was associated with considerable non-haematologic toxicity of mucositis and vomiting, and high HDFL-induced neurotoxicity cast another concern on its safety. A recent report dealt with FOLFIRI regimen in poor performers (Beretta et al, 2006). Its response rate was promising (40%), but grade 3–4 neutropenia (17%) was forcing dose reduction. Therefore, none of these regimens is widely accepted as the reference, and there is a vital need for new regimens that procure dose intensity without impeding safety.
S-1 has been shown to be safe in Korean patients. In the previous phase II trial, patients tolerated the highest dose intensity ever reported, and grade 4 toxicity was not reported (Jeung et al, 2007). Therefore, we hypothesised that S-1 could also be applied in patients whose general condition is compromised. When designing this trial, we adopted a 2-week schedule of S-1 instead of the conventional 4-week schedule. It is based on the knowledge that: (1) S-1-related adverse reactions commonly appear 2–3 weeks after treatment starts (Nagashima et al, 2005); (2) a 2-week schedule showed the possibility of mitigated toxicity and prolonged the medication period (Kimura et al, 2003); (3) this schedule showed feasibility in heavily pretreated metastatic colorectal cancer (Jeung et al, 2006); and (4) recent phase I study of 2-week S-1 schedule demonstrated an antitumour activity in chemotherapy-refractory gastric cancer, with a similar pharmacokinetic profile to the conventional schedule (Zhu et al, 2007).
Another concern in designing this trial was how to measure the efficacy of S-1 in our patients, since few studies have concentrated on this poor performance status population (Wilke et al, 1990; Hsu et al, 1997; Jeung et al, 2007). Gastric cancer is mixed with considerable non-measurable proportion, and it is higher in Korean patients compared with Western populations. Peritoneal metastasis or locoregional recurrence comprises approximately 50% of initial recurrence of gastric cancer, most of which cannot be numerically assessed by conventional imaging (Yoo et al, 2000). Therefore, we concentrated on 1-year survival rather than response rate to measure the clinical benefit of S-1.
Performance status is an indicator of a patient's global ability and it correlates with survival time. Preoperative performance status of ECOG 2–3 in non-curable gastric cancer patients is associated with a 1-year survival rate of 17%, compared with 43% for ECOG 0-1 patients (Maehara et al, 1993). In a prospective trial of 5-FU and cisplatin, ECOG 2–3 patients showed a 1-year survival of 30% (Rougier et al, 1994). Our study was designed to prove a 1-year survival of 30%, which is somewhat high considering the patients' general condition and proportion of prior exposure to chemotherapy. Nevertheless, this survival rate is a currently acceptable range expected from any new regimen aiming at second- or third-line treatment for gastric cancer (Assersohn et al, 2004; Chun et al, 2004; Wilson et al, 2005; Horinaka et al, 2006).
We obtained a 1-year survival rate of 29% with a response rate of 11%. This suggests an activity of S-1 in gastric cancer patients even with poor performance status. Although direct comparison is difficult, our result was comparable to other single-agent chemotherapy regimens in pretreated patients or poor performers (Cascinu et al, 1998; Chun et al, 2004; Horinaka et al, 2006). Moreover, there was no difference in response or treatment compliance according to other poor prognostic factors such as age, comorbidity, or performance status. However, it should be noted that treatment duration and efficacy seems dependent on previous treatment. As first-line treatment, the median treatment cycle was four, and five patients (23%) completed 12 cycles. Five patients (23%) showed an objective response, which was comparable to a conventional phase II trial of S-1 (Jeung et al, 2007). In contrast, S-1 in second- or third-line treatment showed only one response, and the median treatment cycle was two. This implies that although S-1 monotherapy has an activity in poor performance status, this activity appears to be restricted to first-line chemotherapy. There is currently inadequate evidence that S-1 monotherapy could be successfully used for salvage treatment in pretreated cases.
Another point to consider is that we observed a different toxicity profile than that reported in a previous phase II trial (Jeung et al, 2007). Our patients suffered more haematologic and non-haematologic toxicity, and the incidences of grade 3 neutropenia, diarrhoea, and anorexia were increased. It is no wonder if we assume that poor performance status reflects the global deterioration of the host-defense activity against the tumour and treatment burden. Although we did not assess the quality of life in this study, most patients (83%) did not suffer further weight loss during the S-1 treatment, and two patients gained weight and increasing appetite. Moreover, rare grade 4 toxicity and the lack of toxicity-related death indicate that S-1 could be applied safely to this clinical setting. However, our result also indicates that scrupulous evaluation of the adverse reaction needs for these patients is because of the more severe toxicity profile.
To conclude, our prospective study suggested that S-1 monotherapy is tolerable and has modest activity in gastric cancer patients with poor performance status. Owing to the natural history of gastric cancer, patients are particularly prone to clinical deterioration after diagnosis or during treatment course. Therefore, it is especially important to consider these patients to improve the global treatment outcome of gastric cancer.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Andreyev HJN, Norman AR, Oates J, Cunningham D (1998) Why do patients with weight loss have a worse outcome when undergoing chemotherapy for gastrointestinal malignancies? Eur J Cancer 34: 503–509
Assersohn L, Brown G, Cunningham D, Ward C, Oates J, Waters JS, Hill ME, Norman AR (2004) Phase II study of irinotecan and 5-fluorouracil/leucovorin in patients with primary refractory or relapsed advanced oesophageal and gastric carcinoma. Ann Oncol 15: 64–69
Beretta E, Di Bartolomeo M, Buzzoni R, Ferrario E, Mariani L, Gevorgyan A, Bajetta E (2006) Irinotecan, fluorouracil and folinic acid (FOLFIRI) as effective treatment combination for patients with advanced gastric cancer in poor clinical condition. Tumori 92: 379–383
Cascinu S, Graziano F, Cardarelli N, Marcellini M, Giordani P, Menichetti ET, Catalano G (1998) Phase II study of paclitaxel in pre-treated advanced gastric cancer. Anticancer Drugs 9: 307–310
Chao Y, Teng HC, Hung HC, King KL, Li CP, Chi KH, Yen SH, Chang FY (2000) Successful initial treatment with weekly etoposide, epirubicin, cisplatin, 5-fluorouracil and leucovorin chemotherapy in advanced gastric cancer patients with disseminated intravascular coagulation. Jpn J Clin Oncol 30: 122–125
Chun JH, Kim HK, Lee JS, Choi JY, Lee HG, Yoon SM, Choi IJ, Ryu KW, Kim YW, Bae JM (2004) Weekly irinotecan in patients with metastatic gastric cancer failing cisplatin-based chemotherapy. Jpn J Clin Oncol 23: 8–13
Cunningham D, Hole D, Taggart DJ, Soukop M, Carter DC, McArdle CS (1987) Evaluation of the prognostic factors in gastric cancer: the effect of chemotherapy on survival. Br J Surg 74: 715–720
Horinaka S, Zenda S, Boku N, Fukutomi A, Yoshino T, Onozawa Y (2006) Weekly paclitaxel as second-line chemotherapy for advanced or recurrent gastric cancer. Gastric Cancer 9: 14–18
Hsu CH, Yeh KH, Chen LT, Liu JM, Jan CM, Chen YC, Cheng AL (1997) Weekly 24-h infusion of high-dose 5-fluorouracil and leucovorin in the treatment of advanced gastric cancer: an effective and low-toxic regimen for patients with poor general condition. Oncology 54: 275–280
Janunger KG, Halfstrom L, Nygen P, Glimelius B (2001) A systematic overview of chemotherapy effects in gastric cancer. Acta Oncol 40: 309–326
Jeung HC, Rha SY, Cho BC, Yoo NC, Roh JK, Roh WJ, Chung HC, Ahn JB (2006) A phase II trial of S-1 monotherapy in metastatic colorectal cancer after failure of irinotecan- and oxaliplatin-containing regimens. Br J Cancer 95: 1637–1641
Jeung HC, Rha SY, Kim HK, Lim HY, Kim S, Kim SY, Gong SJ, Park CH, Ahn JB, Noh SH, Chung HC (2007) Multi-institutional phase II study of S-1 monotherapy in advanced gastric cancer with pharmacokinetics and pharmacogenomic evaluations. Oncologist 12: 543–554
Kimura Y, Kikkawa N, Iijima S, Kato T, Naoi Y, Hayashi T, Tanigawa T, Yamamoto H, Kurokawa E (2003) A new regimen for S-1 therapy aiming at adverse reaction mitigation and prolonged medication by introducing a 1-week drug-free interval after each 2-week dosing session: efficacy and feasibility in clinical practice. Gastric Cancer 6: S34–S39
Koizumi W, Kurihara M, Nakano S, Hasegawa K (2000) Phase II study of S-1, a novel oral derivative of 5-fluorouracil, in advanced gastric cancer. Oncology 58: 191–197
Lavin PT, Bruckner HW, Plaxe SC (1982) Studies in prognostic factors relating to chemotherapy for advanced gastric cancer. Cancer 50: 2016–2023
Maehara Y, Sugimachi K, Ogawa M, Kakegawa T, Shimazu H, Tomita M (1993) Influence of preoperative performance status on survival time of patients with advanced gastric cancer following noncurative resection. Anticancer Res 13: 201–204
Nagashima F, Ohtsu A, Yoshida S, Ito K (2005) Japanese nationwide post-marketing survey of S-1 in patients with advanced gastric cancer. Gastric Cancer 8: 6–11
Rougier P, Ducreux M, Mahjoubi M, Pignon JP, Bellefqih S, Oliveira J, Bognel C, Lasser P, Ychou M, Elias D, Cvitkovic E, Armand JP, Droz JP (1994) Efficacy of combined 5-fluorouracil and cisplatinum in advanced gastric carcinomas. A phase II trial with prognostic factor analysis. Eur J Cancer 30A: 1263–1269
Sakata Y, Ohtsu A, Horikoshi N, Sugimachi K, Mitachi Y, Taguchi T (1998) Late phase II study of novel oral fluoropyrimidine anticancer drugs S-1 (1 M tegafur-0.4 M gimestat–1 M otastat potassium) in advanced gastric cancer patients. Eur J Cancer 34: 1715–1720
Sugimachi K, Maehara Y, Horikoshi N, Shimada Y, Sakata Y, Mitachi Y, Taguchi T (1999) An early phase II study of oral S-1, a newly developed 5-fluorouracil derivative for advanced and recurrent gastrointestinal cancers. Oncology 57: 202–210
Trumper M, Ross PJ, Cunningham D, Norman AR, Hawkins R, Seymour M, Harper P, Iveson T, Nicolson M, Hickish T (2006) Efficacy and tolerability in elderly patients with advanced oesophago-gastric cancer: a pooled analysis of three clinical trials. Eur J Cancer 42: 827–834
Wilke H, Preusser P, Fink U, Achterrath W, Meyer HJ, Stahl M, Lenaz L, Meyer J, Siewert JR, Geerlings H, Kohne-Wompner CH, Harstrick A, Schmoll HJ (1990) New developments in the treatment of gastric carcinoma. Semin Oncol 17: S61–S70
Wilson D, Hiller L, Geh JI (2005) Review of second-line chemotherapy for advanced gastric adenocarcinoma. Clin Oncol 17: 81–90
Yoo CH, Noh SH, Shin DW, Choi SH, Min JS (2000) Recurrence following curative resection for gastric carcinoma. Br J Surg 87: 236–242
Zhu AX, Clark JW, Ryan DP, Meyerhardt JA, Enzinger PC, Earle CC, Fuchs CS, Regan E, Anbe H, Houghton M, Zhang J, Urrea P, Kulke MH (2007) Phase I and pharmacokinetic study of S-1 administered for 14 days in a 21-day cycle in patients with advance upper gastrointestinal cancer. Cancer Chemother Pharmacol 59: 285–293
Acknowledgements
This work was supported by the Korea Science and Engineering Foundation (KOSEF) Grant funded by the Korean government (MOST) (R11-2000-082-03002-0). This study was supported by the KOSEF through the Cancer Metastasis Research Center at Yonsei University. We are grateful to Eun Young Joo and Seung Ah Kang, clinical research coordinator for their sincere help in clinical trial management and data documentation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Conflict of interest statement
The authors indicated no potential conflicts of interest.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Jeung, HC., Rha, S., Shin, S. et al. A phase II study of S-1 monotherapy administered for 2 weeks of a 3-week cycle in advanced gastric cancer patients with poor performance status. Br J Cancer 97, 458–463 (2007). https://doi.org/10.1038/sj.bjc.6603902
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6603902
Keywords
This article is cited by
-
The relationship between the number of examined lymph nodes and the efficacy of chemotherapy for gastric cancer
Surgery Today (2020)
-
Gastric cancer with metastases to the scaphoid: a case report
Journal of Orthopaedic Science (2015)
-
Phase II and UGT1A1 genotype study of irinotecan dose escalation as salvage therapy for advanced gastric cancer
British Journal of Cancer (2012)
-
Phase II trial of S-1 monotherapy in elderly or frail patients with metastatic colorectal cancer
Investigational New Drugs (2011)
-
Phase II study of S-1 monotherapy in paclitaxel- and cisplatin-refractory gastric cancer
Cancer Chemotherapy and Pharmacology (2009)
