
Abstract
In children, treatment regimen for high-grade gliomas (HGG) and diffuse intrinsic pontine gliomas (DIPG) are generally not stratified according to disease stage. The hypothesis was that secondary disseminating disease (SDD) in children with HGG is related to an even worse outcome. Description of SDD pattern was performed. In total, 270 children with newly diagnosed HGG or DIPG were eligible for retrospective analysis of SDD. Medical and computer records of these patients were reviewed for demographic characteristics, sites of dissemination, prognostic variables. Forty-six (17%) of the 270 patients had developed SDD. The median time to SDD was 8.2 months. The median overall survival (OS) after dissemination was 3.2 months. The SDD was located parenchymal in the supratentorial (34.8%), infratentorial (6.5%), supratentorial and infratentorial (19.6%), spinal (10.9%), spinal and cerebral (6.5%) regions of the CNS, or leptomeningeal (21.7%). For HGG patients, the median OS was shorter among patients with SDD than among patients without SDD (1.02 vs 1.41 years, P=0.0495). In the group of patients with SDD, patients with cerebrospinal fluid dissemination had a worse outcome compared with patients with parenchymal metastases. Summarising, SDD is a negative prognostic factor for patients with HGG outside the pons. Treatment stratification should be considered.
Similar content being viewed by others


Main
Tumours of the central nervous system (CNS) are the most common solid neoplasms in children (Pollack, 1994). High-grade gliomas (HGG) (World Health Organization grade III and IV) constitute only 10–20% of childhood CNS tumours. In the German Database for HGG, about 40% of children were registered with diffuse intrinsic pontine glioma (DIPG). The diagnosis of DIPG is performed by radiological criteria: larger than 50% of the pontine diameter (Wagner et al, 2006) combined normally a short history of disease. All other criteria are not suitable to distinguish between focal and diffuse pontine glioma: DIPG includes tumours of WHO grade I-IV histology showing contrast enhancement on magnetic resonance imaging (MRI) and areas of necrosis and /or exophytic components.
Owing to the small number of children with these neoplasms, it is difficult to design and conduct prospective clinical trails of treatments for HGG. In contrast to other childhood malignancies, where treatment according to disease stage is standard, HGG patients with metastases are treated the same way as patients with localised disease. Owing to low incidences of secondary metastases in HGG and DIPG there is no impact for alternative treatment strategies.
In a previous study, we showed that about 3% of children with HGG or DIPG initially present with disseminated tumour spread at diagnosis so called primary dissemination of disease, stressing the need for a complete initial diagnostic workup, including neuroaxis MRI and cerebrospinal fluid (CSF) examination (Benesch et al, 2005). Most reports on tumour dissemination of HGG and DIPG after diagnosis, so called secondary dissemination of disease (SDD), are case reports or small series; few reports include large numbers of patients (Packer et al, 1985, 1990, 1993; Dropcho et al, 1987; Vertosick and Selker, 1990; Grabb et al, 1992; Arita et al, 1994; Heideman et al, 1997; Donahue et al 1998; Allen et al, 1999; Endo et al, 2003; Saito et al, 2003). These series mostly come from the late 1980s and early 1990s, when MRI was not routinely available for follow-up investigations (Awad et al, 1986; Vertosick and Selker, 1990; Arita et al, 1994). Secondary disseminating disease has been reported in 14–25% of adult patients with HGG or DIPG. The percentage of paediatric patients with SDD varies remarkably among studies (Table 1).
Data of patients with HGG and DIPG included into this retrospective study were recruited from a German HGG database in which patients were registered since 1995 in a prospective series of consecutive single-arm protocols using common diagnostic criteria for eligibility, with overall survival (OS) as the primary end point for comparison. The induction treatments consisted of conformal normal-fractionated radiotherapy and chemotherapy (Wolff et al, 2000, 2002a, 2002b, 2002c; Wagner et al, 2005), such as trofosfamide and oral etoposide (Dropcho et al, 1987; Vertosick and Selker, 1990); cisplatin, etoposide, ifosfamide (PEI) (Wolff et al, 2002b); or PEI plus vincristine or PEI plus high-dose methotrexate (Wagner et al, 2005). Data on metastatic disease were included in baseline information, but they did not alter the treatment strategies. In addition, all patients who developed disease relapse or progression were offered treatment according to the same protocol (Wagner et al, 2004). The most recent protocol treatment for patients with relapsed disease differed for those with completely vs incompletely resected recurrent disease, but the presence of metastases did not alter these treatment recommendations.
We performed a retrospective study in children with HGG or DIPG to determine, whether SDD is related to a worse outcome. Description of the pattern of SDD and the impact of SDD on OS comparing HGG with DIPG was a secondary objective; in order to obtain reliable data for treatment stratification in future studies.
Materials and methods
Only patients with reliable radiographic and/or histopathologic evidence of SDD or non-SDD were included in this analysis. Eligibility criteria included informed consent of parents or legal guardians. Secondary disseminating disease was defined as the unequivocal presence of SDD outside the area of the primary tumour that had been documented by at least two different sources of information (e.g., two follow-up MRI studies) or proven by autopsy. Patients who were registered but who did not follow treatment guidelines were not excluded. All computer records and paper records were reviewed for diagnostic criteria of SDD by two investigators (MB, SW). Referring hospitals were contacted for missing data.
Treatment
Patients of this study were recruited of a database in which patients were registered from July1995 until March 2005. Totally 653 patients were registered, and were enrolled into sequential protocols (A–D) of the study for HGG and DIPG of the Paediatric Oncology and Hematology Society of the German Language Group, initiated by JW. All protocols were reviewed and approved by Deutsche Krebsgesellschaft and the ethics committees of the participating centres.
Treatment protocols A (1995–1997), B (1997–1999), C (1999–2003) and D (has started 2003) have been previously described (Wolff et al, 2000, 2002a; Wagner et al, 2005). In all protocols, patients underwent conventional fractionated radiotherapy after surgery with a total dose of 59.6 or 54.0 Gy for children <6 years old and for children with brainstem gliomas. The volume of tissue irradiated was the tumour volume, as determined on preoperative MRI, plus a 2-cm margin. One year chemotherapy in protocol A consisted of oral trofosfamide (100 mg m−2 per day) and oral etoposide (25 mg m−2 per day) given once daily for 21 days, repeated after 1 week of rest. In the following protocol (B), patients underwent an intensive synchronous radiochemotherapy regimen soon after surgery, consisting of two courses of induction chemotherapy (cisplatin and etoposide in the first and cisplatin, etoposide and ifosfamide in the second course). This synchronous radiochemotherapy was also used for protocols C and D. In protocol C, vincristine (1.5 mg m−2) was added once a week during synchronous radiochemotherapy. In addition, six courses of PEI were given as consolidation chemotherapy every 4 weeks after. In protocol D, patients were either randomised to receive two doses of high-dose methotrexate (5 g m−2) before induction radiochemotherapy or to immediately undergo induction radiochemotherapy. Consolidation therapy in this study consisted of lomustine, vincristine and prednisone (every 6 weeks for eight courses).
Response to treatment was defined as previously described (Gnekow 1995; Wolff et al, 2000). Follow-up MRI was performed every 3–6 months after radiochemotherapy according to protocol guidelines and when the clinical course suggested disease progression.
Diagnostic evaluation
The initial diagnostic evaluation for entering the study included a complete physical and detailed neurological examination as well as a brain MRI study with and without gadolinium. Spinal MRI and cytologic examination of CSF were recommended but not mandatory. Histopathologic diagnosis and grading of tumours were performed by local neuropathologists according to the World Health Organization classification of CNS tumours (Kleihues et al, 1993). Central neuropathologic review of the local pathological diagnosis was performed in almost all patients at the Neuropathological Reference Centre at the University of Bonn in Germany. The radiological diagnosis of a DIPG was reviewed by the Neuroradiological Reference Centre in Würzburg, Germany. The criteria for DIPG were that the tumour had to be larger than 50% of the pontine diameter. Criteria for HGG were a histopathological diagnosis of a WHO°III or °IV glioma. The extent of surgical resection was documented using the neurosurgeon's report and postoperative contrast-enhanced MRI. Diagnosis of SDD was based on MRI studies and was defined as evidence of linear or nodular contrast-enhancing lesions outside the area of the primary tumour on follow-up MRI studies.
Statistical analysis
Statistical analyses were performed using the commercially available program SPSS version 12.0; SPSS Inc., Chicago, IL, USA). The χ2-test was used to compare patient subgroups concerning HGG, DIPG, SDD, age, gender, extent of resection. The Kaplan–Meier method was used to estimate OS rates and median OS time. The 95% confidence interval (CI) was defined by the program as the 95% probability that the calculated OS time can be found within this interval. Differences in OS were analysed with the log-rank test and were estimated as relevant different with a P-value of <0.05. Results were updated 30 April 2005.
Results
Patient characteristics
During the period studied, 653 patients <21 years old with newly diagnosed HGG or DIPG were registered into the above described database. Of the 653 patients, 368 patients had HGG and 257 had DIPG. We were not able to classify the remaining 28 patients because of the lack of a reference radiological report and not knowing the extent of the tumour. The eligibility criteria for this study were met by 270 patients. Of these, 146 had HGG and 110 had DIPG. Fourteen patients could not be classified into either of these groups because of a lack of clinical data. Forty-six (17%) of the 270 patients developed SDD during the period studied. All patients did not have metastases at primary diagnosis. The clinical features of these patients are summarised in Table 2. The overall incidence of SDD was 12.7% (14/110) for patients with DIPG, and 21.9% (32/146) for patients with HGG.
The median time between diagnosis of the primary tumour and SDD was 8.2 months (range, 2.2 months–6.2 years) for the whole group, 7.2 months (range, 4.6 months–2.2 years) for the DIPG group, and 8.2 months (range, 2.2 months–6.2 years) for the HGG group. In the majority of patients (n=33; 71%), metastases occurred soon after initial diagnosis (i.e., within 1 year). In six patients (13%) metastases were observed between 1 and 2 years after initial diagnosis, and in five patients (11%), metastases occurred more than 2 years after diagnosis (no data available for 2 patients, 5%).
CSF examination was initially performed in 11 patients and was positive in two of these patients. Metastases were found at various combinations of anatomic sites in various combinations (Table 3). Extra-CNS metastases were not found.
Primary treatment and response
The initial treatment that the 46 patients with SDD received is presented in Table 2. The number of patients treated according to protocols A–D was as follows: protocol A, n=7 (15%); protocol B, n=11 (24%); protocol C, n=20 (44%) and protocol D, n=8 (17%). Eight weeks after the initiation of therapy, response was evaluated in the 36 patients before the onset of SDD without a complete tumour resection (Table 4), revealing a complete remission in two patients (6%), partial remission in six patients (17%), stable disease in 20 patients (56%) and progressive disease in eight patients (22%). Six of the seven patients who had complete tumour resections were in continuous complete remission at the time of the first response evaluation. Data on remission status were not available for three patients.
Data on the systemic treatment of patients with SDD were available for only 30 patients because this was no further a protocol question in the A–D protocol (Table 3). Unfortunately, data on second surgeries were not uniformly reported.
OS of patients with and without SDD
Evaluation of all 270 patients for whom SDD data were available did not reveal any difference in OS between patients with and without SDD. The median OS was 1.02±0.14 years (95% CI, 0.75–1.30 years) for patients with SDD and 1.01±0.06 years (95% CI, 0.90–1.12 years) (P=0.473) for patients without SDD. For the patients with HGG, we found a survival difference between patients with and without SDD. The median OS of HGG patients with SDD (n=32) was 1.02±0.21 years (95% CI, 0.61–1.43 years), and the median OS of HGG patients without SDD (n=14) was 1.41±0.28 years (95% CI, 0.86–1.95 years) (P=0.0495). In contrast, in the subgroup of patients with DIPG, no differences in median OS were found between patients with and without SDD.
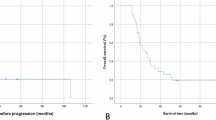
Further analyses of HGG patients revealed a difference in median OS between patients with (n=15; median OS=0.86±0.2 years; 95% CI, 0.46–1.25 years) and without (n=42; 2.28±0.95 years; 95% CI, 0.42–4.15 years) SDD among those with grade III tumours (P=0.001, Figure 1A) but not among those with grade IV tumours (Figure 1B). Similarly, a difference in OS between HGG patients with (n=20; median OS=0.79±0.10 years; 95% CI, 0.59–0.99 years) and without (n=73; median OS=1.37±0.15 years; 95 % CI, 0.8–1.66 years; P=0.023) SDD was found in younger children (< 13 years) but not in older children (⩾13 years).

Kaplan–Meier curves for OS of high-grade glioma grade III patients (A) and grade IV patients (B) with and without SDD.
Overall survival was longer for patients with parenchymal metastases (n=22; median OS for patients without leptomeningeal/ventricular SDD=1.18±0.19 years; 95% CI, 0.80–1.56 years) compared with patients whose tumours had spread through the CSF (n=22; median OS of patients with leptomeningeal/ventricular SDD=0.72±0.05 years; 95% CI, 0.62-0.81 years) (P=0.049) (Figure 2). For patients with SDD, the median OS after SDD was 0.27±0.05 years (n=44, 95% CI, 0.18–0.37 years) for the HGG and DIPG groups combined. No patient survived longer than 1.8 years after the diagnosis of SDD; the 1-year OS rate after being diagnosed with SDD was 16.7±6.1%. A statistically significant difference in OS was found when the different sites of the intracranial metastases were compared.

Kaplan–Meier curves for OS of HGG and DIPG patients with secondary disseminated disease with and without leptomenigeal (LM)/ventricular dissemination.
Discussion
This is the largest survey on SDD in paediatric HGG and DIPG patients to date. As expected, the majority (72%) of cases of SDD were detected soon after initial diagnosis (within 1 year). However, there was an interesting small group of five patients who developed SDD more than 2 years after diagnosis, indicating that the biological characteristics of HGG and DIPG tumours are more complex than anticipated.
In our study, SDD did influence OS in patients with HGG, as they had a shorter OS than those without SDD. However, SDD had little impact on the OS of the HGG and DIPG groups combined. Secondary disseminating disease did not reduce OS in the DIPG group, suggesting that rapid local tumour progression was the main cause of the short OS in these patients and that SDD had no additional effect. We conclude, therefore, that in DIPG patients, treatment should primarily focus on controlling local disease.
Among patients with HGG, OS was shorter in patients with SDD in grade III tumours and with age of <13 years. We suggest that HGG patients in these subgroups might benefit from an intensive treatment of metastases, such as spinal or local irradiation or surgery, since tumour progression at the primary site seems to occur less rapidly compared to patients with DIPG.
The overall incidence of SDD in our study was 17% (HGG, 22%; DIPG, 13%), which is in the lower part of the range reported by others (Table 1, Dropcho et al, 1987). The highly variable incidence rates of SDD in these other studies might be explained by the fact that they were single-centre-based studies with a limited number of patients, or that they had incomplete CNS staging and different MRI surveillance strategies. In addition, treatment intensity, including high-dose chemotherapy, and post-mortem examinations might have had some impact on the proportion of patients with HGG or DIPG who were diagnosed with SDD in those studies (Table 1). Treatment intensity, in particular, might alter the metastatic potential of HGG and DIPG, thereby affecting the OS in patients with these diseases. Normally data on SDD in HGG or DIPG patients were seldom available because the patients had a short OS, so dissemination did not have time to occur.
Interestingly, in HGG and DIPG patients, dissemination to leptomeningeal and ventricular tumour sites resulted in shorter OS compared with parenchymal metastases; this observation could redirect new treatment approaches. One of these new approaches could be intrathecal administration of chemotherapeutic drugs as it was proposed by Donahue et al (1998).
Before initiating new treatment approaches, however, better diagnostic standards, such as craniospinal MRI and mandatory CSF examination, must be implemented in addition to more sensitive methods for screening CSF. Risk factor analysis (e.g., genetic factors to predict SDD in HGG) is another important issue to be addressed in future studies. Even if this retrospective study is the largest survey of patients with SDD, patient numbers are still small. In order to get more experiences with SDD in the group of HGG and DIPG, new protocols should at least plan prospective detailed documentation of SDD. For the first time it was reported that SDD is as a negative prognostic factor in HGG. This finding should still be confirmed by other studies, however, it does suggest a biological relevance of secondary metastases in high-grade glioma, and leads to reconsider intrathecal therapy for these tumours in children.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Allen J, Siffert J, Donahue B, Nirenberg A, Jakacki R, Robertson P, DaRosso R, Thoron L, Rosovsky M, Pinto R (1999) A phase I/II study of carboplatin combined with hyperfractionated radiotherapy for brainstem gliomas. Cancer 86: 1064–1069
Arita N, Taneda M, Hayakawa T (1994) Leptomeningeal dissemination of malignant gliomas. Incidence, diagnosis and outcome. Acta Neurochir (Wien) 126: 84–92
Awad I, Bay JW, Rogers L (1986) Leptomeningeal metastasis from supratentorial malignant gliomas. Neurosurgery 19: 247–251
Benesch M, Wagner S, Berthold F, Wolff JEA (2005) Primary dissemination of high-grade gliomas in children: experiences from four studies of the Pediatric Oncology and Hematology Society of the German Language Group (GPOH). J Neurooncol 72: 179–183
Donahue B, Allen J, Siffert J, Rosovsky M, Pinto R (1998) Patterns of recurrence in brain stem gliomas: evidence for craniospinal dissemination. Int I Radiat Oncol Biol Phys 40: 670–680
Dropcho EJ, Wisoff JH, Walker RW, Allen JC (1987) Supratentorial malignant gliomas in childhood: a review of fifty cases. Ann Neurol 22: 355–364
Endo H, Kumabe T, Jokura H, Shirane R, Ariga H, Takai Y, Yoshimoto T (2003) Leptomeningeal dissemination of cerebellar malignant astrocytomas. J Neurooncol 63: 191–199
Gnekow A (1995) Recommendations of the brain tumor subcommittee for reporting of trials. Med Pediatr Oncol 24: 104–108
Grabb PA, Albright AL, Pang D (1992) Dissemination of supratentorial malignant gliomas via the cerebrospinal fluid in children. Neurosurgery 30: 64–71
Heideman RL, Kuttesch Jr J, Gajjar AJ, Walter AW, Jenkins JJ, Li Y, Sanford RA, Kun LE (1997) Supratentorial malignant gliomas in childhood. Cancer 80: 497–504
Kleihues P, Burger PC, Scheithauer BW (1993) Histological Typing of Tumours of the Central Nervous System 2nd edn, Berlin: Springer-Verlag
Packer RJ, Allen JC, Goldwein JL, Newall J, Zimmerman RA, Priest J, Tomita T, Mandelbaum DE, Cohen BH, Finlay JL, Wara WM (1990) Hyperfractionated radiotherapy for children with brainstem gliomas: a pilot study using 7200 cGy. Ann Neurol 27: 167–173
Packer RJ, Boyett JM, Zimmerman RA, Rorke LB, Kaplan AM, Albright AL, Selch MT, Finlay JL, Hammond GD, Wara WM (1993) Hyperfractionated radiation therapy (72 Gy) for children with brain stem gliomas. A Children's Cancer Group Phase I/II Trial. Cancer 72: 1414–1421
Packer RJ, Siegel KR, Sutton LN, Litmann P, Bruce DA, Schut L (1985) Leptomeningeal dissemination of primary central nervous system tumors of childhood. Ann Neurol 18: 217–221
Pollack IF (1994) Brain tumors in children. New Engl J Med 331: 1500–1507
Saito R, Kumabe T, Jokura H, Shirane R, Yoshimoto T (2003) Symptomatic spinal dissemination of malignant astrocytoma. J Neurooncol 61: 227–235
Vertosick Jr FT, Selker RG (1990) Brain stem and spinal metastases of supratentorial glioblastoma multiforme: a clinical series. Neurosurgery 27: 516–521
Wagner S, Erdlenbruch B, Langler A, Gnekow A, Kuhl J, Albani M, Volpel S, Bucsky P, Emser A, Peters O, Wolff JE (2004) Oral topotecan in children with recurrent or progressive high-grade glioma: a Phase I/II study by the German Society for Pediatric Oncology and Hematology. Cancer 100: 1750–1757
Wagner S, Reinert C, Schmid HJ, Liebeskind AK, Jorch N, Längler N, Graf N, Warmuth-Metz M, Pietsch T, Peters O, Wolff JEA (2005) High-dose methotrexate prior to simultaneous radiochemotherapy in children with malignant high-grade gliomas. Anticancer Res 25: 2583–2587
Wagner S, Warmuth-Metz M, Emser A, Gnekow A, Sträter R, Rutkowski S, Jorch N, Schmid HJ, Berthold F, Graf N, Kortmann RD, Pietsch T, Sörensen N, Peters O, Wolff JEA (2006) Treatment options in childhood pontine. J Neuro-Oncol 79: 281–287
Wolff JE, Gnekow AK, Kortmann RD, Pietsch T, Urban C, Graf N, Kuhl J (2002c) Preradiation chemotherapy for pediatric patients with high-grade glioma. Cancer 94: 264–271
Wolff JE, Molenkamp G, Westphal S, Pietsch T, Gnekow A, Kortmann RD, Kuehl J (2000) Oral trofosfamide and etoposide in pediatric patients with glioblastoma multiforme. Cancer 89: 2131–2137
Wolff JE, Westphal S, Molenkamp G, Gnekow A, Warmuth-Metz M, Rating D, Kuehl J (2002a) Treatment of paediatric pontine glioma with oral trophosphamide and etoposide. Br J Cancer 87: 945–949
Wolff JEA, Wagner S, Sindichakis M, Pietsch T, Gnekow A, Kortmann R-D, Sträter R, Kühl J (2002b) Simultaneous radiochemotherapy in pediatric patients with high-grade glioma: a phase I study. Anticancer Res 22: 3569–3572
Acknowledgements
Supported by the Deutsche Kinderkrebsstiftung in Germany.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Wagner, S., Benesch, M., Berthold, F. et al. Secondary dissemination in children with high-grade malignant gliomas and diffuse intrinsic pontine gliomas. Br J Cancer 95, 991–997 (2006). https://doi.org/10.1038/sj.bjc.6603402
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6603402
Keywords
This article is cited by
-
Advances in Treatment of Diffuse Midline Gliomas
Current Neurology and Neuroscience Reports (2023)
-
Disseminated diffuse midline glioma associated with poorly differentiated orbital lesion and metastases in an 8-year-old girl: case report and literature review
Child's Nervous System (2022)
-
Leptomeningeal and subependymal seeding of diffuse intrinsic pontine glioma: a case report
Child's Nervous System (2022)
-
How ten-years of reirradiation for paediatric high-grade glioma may shed light on first line treatment
Journal of Neuro-Oncology (2022)
-
Adult brainstem glioma: a multicentre retrospective analysis of 47 Italian patients
Neurological Sciences (2021)
