
Abstract
The goal of this phase I study was to develop a novel schedule using oral etoposide and infusional topotecan as a continually alternating schedule with potentially optimal reciprocal induction of the nontarget topoisomerase. The initial etoposide dose was 15 mg m−2 b.i.d. days (D)1–5 weeks 1,3,5,7,9 and 11, escalated 5 mg per dose per dose level (DL). Topotecan in weeks 2,4,6,8,10 and 12 was administered by 96 h infusion at an initial dose of 0.2 mg m−2 day−1 with a dose escalation of 0.1, then at 0.05 mg m−2 day−1. Eligibility criteria required no organ dysfunction. Two dose reductions or delays were allowed. A total of 36 patients with a median age of 57 (22–78) years, received a median 8 (2–19) weeks of chemotherapy. At DL 6, dose-limiting toxicities consisted of grade 3 nausea, vomiting and intolerable fatigue. Three patients developed a line-related thrombosis or infection and one subsequently developed AML. There was no febrile neutropenia. There were six radiologically confirmed responses (18%) and 56% of patients demonstrated a response or stable disease, typically with only modest toxicity. Oral etoposide 35 mg m−2 b.i.d. D1–5 and 1.8 mg m−2 96 h (total dose) infusional topotecan D8–11 can be administered on an alternating continual weekly schedule for at least 12 weeks, with promising clinical activity.
Similar content being viewed by others

Main
Etoposide and topotecan are both schedule-dependent cytotoxics that have a broad spectrum of antineoplastic activity. Both interact with topoisomerases, essential nuclear enzymes that cleave DNA to reduce torsional strain during replication and recombination. Topotecan interacts with topoisomerase I (topo I), inducing reversible single-stranded breaks and a compensatory upregulation in topoisomerase II (topo II). (Liu, 1989) Etoposide binds to topo II to cause double-stranded DNA breaks with a compensatory increase in topo I. This see-saw compensation occurs with a delay such that sequential treatment may offer the optimal schedule of treatment. (Eder et al, 1998) Continual exposure to schedule-dependent cytotoxics was first shown to be effective for acute lymphocytic leukaemia of childhood and since then, the investigation of protracted exposure to such agents has been the centre of much of the investigation of the pharmacodynamics of chemotherapy. (Furman et al, 1976; Collins et al, 1990). With the positive experience of sequencing the two topoisomerase interacting agents, doxorubicin and topotecan (Seiden et al, 2002), this study was conceived, building on the anticipation that continual exposure may maximise the benefit of repeatedly alternating topoisomerase I and II interacting drugs, and to define the maximum-tolerated dose (MTD).
DNA topoisomerases are nuclear enzymes essential for DNA replication, RNA transcription, chromosomal condensation and mitotic chromatid separation (Caron and Wang, 1993; Gupta et al, 1995; Kohn and Pommier, 2000). There are two major classes of topoisomerases, I and II, in eukaryotic cells. Topoisomerase I relaxes DNA by forming a covalent bond with the 3′-terminus of a DNA nucleotide, producing a single-stranded break (Gupta et al, 1995). Topoisomerase II functions as a dimer and forms a double-stranded cleavage with each topoisomerase II molecule covalently bound to the 5′-terminus of a DNA nucleotide, producing a double-stranded break (Holm et al, 1985). Topoisomerase-targeted agents stabilise a transient covalent enzyme–DNA complex, which produces DNA strand cleavage and apoptosis (Kohn and Pommier, 2000). Preclinical study of the in vivo therapeutic effect of sequential combinations of topoisomerase I- and II-acting drugs in a murine tumour model system reveals that topoisomerase I mRNA and protein levels in the tumour decrease, whereas topoisomerase II mRNA and protein levels rise, after treatment with camptothecins (Eder et al, 1998) and topotecan (Liebes et al, 1998). The reverse effect, a fall in topoisomerase II and concurrent rise in topoisomerase I levels with a topoisomerase II-active drug treatment, is observed, with the suggestion that sequentially combined topoisomerase I and II agents may result in greater than additive tumour cytotoxicity with relatively little increase in toxicity. In vitro studies suggest topoisomerase IIα is cell cycle phase dependent, being only present in the S phase and G2–M phases of the cell cycle in contrast to topoisomerases I and IIβ, which are expressed constitutively throughout the cell cycle (Heck and Earnshaw, 1986; Liu, 1989). Agents interacting with any topoisomerase exhibit cell cycle cytotoxic specificity. However, there is considerable variation, dependent on dose and schedule, with notable differences between in vitro and in vivo observations (Kaufmann, 1991).
The first clinical trials combining topoisomerase I and II inhibitors have shown substantial, albeit nonlethal, toxicity (Ando et al, 1997). These studies have focused on etoposide as the topoisomerase II active agent and have used the camptothecin analogue first. Sequential treatment appeared to have synergistic effects with a suggestion of greater than additive tumour cytotoxicity and no increase in host toxicity. In Seiden et al's study, the alternative combination of doxorubicin and topotecan was chosen because of in vitro observations that firstly, a sequence of doxorubicin (4 days) followed by camptothecin (4 days) produced the greatest tumour growth delay in a murine breast cancer cell line (>50%) with no increase in toxicity and secondly, changes in topoisomerase I levels recovered more rapidly than topoisomerase II levels (Eder et al, 1998; Seiden et al, 2002).
A phase I trial of sequentially administered etoposide and topotecan was therefore constructed on these observations. The objectives of the study were to determine the dose-limiting toxicities (DLTs) and MTD of sequential etoposide (days (D)1–5) and topotecan by 96 h CIV (D8–11) q 14 in patients with refractory solid tumour malignancies.
Patients and methods
Eligibility criteria
The study protocol was reviewed and approved by the Dana Farber/Partners CancerCare (Boston, MA, USA) Institutional Review Board. An informed consent document satisfying all federal and institutional requirements was read by the patients and signed as a condition of their registration. Patients had histologically documented metastatic or inoperable malignant solid tumours, for which there was no known curative or standard palliative therapy. Performance status was Eastern Cooperative Oncology Group 0–2. Patients had adequate hepatic, renal and haematological function determined by a serum glutamate-oxalo-acetate transaminase <2.5 × upper limit of normal, bilirubin <1.5 × upper limit of normal, creatinine clearance >50 ml min−1, an ANC >1500 μl−1 and platelets >150 000 μl−1. Patients had to be more than 3 weeks from last chemotherapy, 2 weeks from surgery, 6 weeks from nitrosoureas or radiotherapy and could not have had prior pelvic radiotherapy. Women of childbearing potential could not be pregnant or lactating and fertile participants had to practise adequate contraception. Additional inclusion criteria included age >18 years and having central venous access; patients had to be capable of taking oral medication with an anticipated life expectancy in excess of 2 months. Eligibility tests had to be performed within 21 days of commencing therapy.
Treatment plan
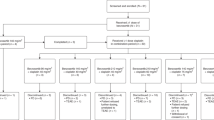
The treatment schema is illustrated in Figure 1. Patients had a history, physical examination, complete blood count and serum chemistries (including liver function tests, Mg2+ and creatinine) performed before each week of therapy, and complete blood count with differential was performed twice each week. These assessments were repeated at 1 month after the last course of treatment and on apparent progression. Documentation of all measurable disease by examination and any appropriate imaging studies (e.g. plain radiograph, computerised tomography and nuclear medicine scan) and EKG were performed before therapy, at week 7 and 13.

Treatment schema. Legend: M, Monday; T, Tuesday; W, Wednesday; Th, Thursday; F, Friday; S, Saturday and S, Sunday. Etoposide administered orally as the i.v. preparation.
On weeks 1, 3, 5, 7, 9 and 11, etoposide was administered orally per allocated dose level (DL) as 10 doses in 5 days with b.i.d. dosing using the i.v. preparation in sterile prefilled syringes. The i.v. preparation of etoposide was used instead of the 50 mg softgel capsules (VePesid®), to allow the necessary dose increments required for the study. Patients were instructed to squirt the contents of a syringe into orange juice and drink it, and store the etoposide at room temperature. On weeks 2, 4, 6, 8, 10 and 12, topotecan was administered by 96-h i.v. infusion as per allocated DL. The 96-h i.v. infusion was chosen to allow ambulatory administration of chemotherapy rather than attending for the standard D1–5 30-min infusions of chemotherapy. Growth factors were not allowed to maintain dose intensity. Etoposide (VePesid™; Bristol-Myers Squibb, Princeton, NJ, USA) and topotecan (Hycamptin™; Smith Kline Beecham, King of Prussia, PA, USA) were prepared from commercially available supplies, formulated and administered per institutional guidelines. Antiemetic therapy could include prn lorazepam 0.5–2.0 mg i.v. and/or prochlorperazine 10 mg p.o. or perphenazine 4 mg p.o. as per institutional standards. The administration of glucocorticoids or 5HT3 antagonists as antiemetics was permitted only after the failure of other antiemetic agents. All patients were treated with warfarin 1 mg p.o. as prophylaxis against line-related thrombosis unless fully anticoagulated for another indication.
Initial DL was 15 mg m−2 b.i.d. etoposide D1–5 with topotecan D8–12 at 0.2 mg m−2 day−1 q 14. Etoposide was then increased in subsequent DLs in increments of 5 mg m−2 dose−1 and topotecan was increased to 0.3 mg m−2 day−1 for DL 2 and then in 0.05 mg m−2 day−1 increments. Initially, three patients fully evaluable were to be recruited to DL 1. If there was no DLT, then accrual continued until the MTD was defined. Dose-limiting toxicities were defined as Gr III neutropenia for >72 h, Gr III thrombocytopenia and Gr III nonhaematological toxicity by CTC 2.0 criteria. Subsequent cohorts of patients were accrued if DLT was not observed in preceding patients over the first 4 weeks of treatment. If DLT was observed, two further patients were enrolled to that DL and the occurrence of a second DLT in 2–6 patients established the previous dose as the MTD. In total, 10 additional patients were treated at the MTD to better define the profile of toxicity at this dose.
Dose modifications were not allowed during the first four weeks of therapy, during which time DLT was evaluated. After week 4, dose modifications were allowed and based on the neutrophil and platelet counts on the first and the fourth day of each treatment week. If the ANC was <1000 μl−1 or platelet count was <100 000 μl−1 on D1, no treatment was given that week and a 25% dose reduction was required for either drug if the ANC was 1000–1499 μl−1 or platelet count was 100 000–149 999 μl−1. Treatment was stopped on D4 of each week of treatment if the ANC was <1499 μl−1 or platelet count was <149 999 μl−1.
Therefore, as the day 4 counts were available after 7/10 doses of etoposide and approximately 75/96 h of topotecan, discontinuation of treatment on day 4 constituted a total dose reduction for that cycle of approximately 44%. Patients requiring more than one dose reduction for a given drug were removed from the study. For the 10 additional patients treated at the MTD, dose reductions were allowed during the first 4 weeks of treatment at the discretion of the treating physician. There were no planned dose escalations.
Disease response criteria
For disease measurable radiologically or by examination, response to therapy was defined in the following manner by standard WHO criteria. A complete response required the disappearance of all measurable disease. For patients with ovarian cancer, a serum CA-125 concentration <35 U l−1 for a minimum of 30 days was also required. Partial response was a reduction in tumour burden of 50% or greater for a minimum of 30 days and decrease in serum CA-125 by at least 50% for patients with an elevated marker. Progressive disease was indicated by a greater than 25% increase in tumour burden or serum tumour marker or the appearance of any new lesion. Stable disease was defined as a decrease in tumour burden or serum tumour marker level not meeting partial response criteria or an increase that did not constitute progressive disease. The following definitions were employed in situations where disease could not be measured by radiological techniques. A complete response required normalisation of tumour marker and complete resolution of evaluable disease such as pleural fluid or ascites, if present. Partial response was a greater than 50% decrease in serum tumour marker, with a reduction in ascites or pleural effusion. A confirmed rising serum tumour marker, even without confirmation by radiological or physical examination, was considered progressive disease.
Results
Between October, 1999 and August, 2002, 36 patients were enrolled. A total of 287 weeks of chemotherapy were delivered (median 8 (2–19) weeks).
Patient characteristics
The characteristics of the 36 patients enrolled into the study are summarised in Table 1. The median age was 57 years (range, 22–78). All patients were evaluable for toxicity. In all, 27 patients had ovarian cancer, four had sarcoma, three had non-small-cell lung cancer and one each had thymoma and cholangiocarcinoma. All patients had progression of tumour with at least one prior chemotherapy regimen, and 16 patients had received more than two prior chemotherapy regimens. Only two patients had ECOG performance score 2.
Additional patients were enrolled to DLs 1 and 2 to replace patients who developed rapid disease progression. One patient developed small bowel obstruction at week 3, one patient developed symptomatic progression of brain metastases at week 3. At DL 4, the study seemed to be near defining the MTD with haematologic toxicity-triggered dose reductions. However, these prevented significant toxicity and the protocol was revised to mandate no dose reductions during the first 4 weeks of therapy. Despite this, DL 4 appeared tolerable and the dose was escalated after a total of nine patients had been enrolled at this DL.
Toxicity
Haematologic and nonhaematologic toxicity are recorded in Tables 1, 2 and 3. Grade 4 haematologic toxicity did not occur, despite continual chemotherapy, effectively prevented by the dose delay and reduction rules. No patient required hospitalisation or antibiotics for neutropenia and there was no febrile neutropenia. Significant nonhaematological toxicity was relatively infrequent. Mild alopecia was common. Six patients experienced mild (one grade 2) infusion catheter-related discomfort or swelling. One patient had a problematic paraneoplastic plexopathy, making evaluation of line-related symptoms difficult, and two patients developed catheter-related infection, both managed conservatively with outpatient oral antibiotic therapy with preservation of the line. Only one catheter-related thrombosis was documented. Although mucositis was notably rare, an unpleasant metallic taste was prominent during treatment with etoposide. At DL 6, continuous nausea, vomiting and fatigue were intolerable. One patient with primary peritoneal cancer and a past history of endometrial cancer subsequently developed AML with normal cytogenetics.
Tumour response
A total of 34 patients were evaluable for response (Table 4). Two were inevaluable because of the interval development of small bowel obstruction and cerebral metastases, after 2 and 3 weeks of therapy, respectively. Overall, six of 34 patients' (18%) tumours demonstrated a response, all but one of which were confirmed 1 month later on imaging, and one of which was a complete response. The duration of response was a median 8.1 months (range 6.8–21.5 months). Tumours in many patients developed minor responses and a response or stable disease was observed in 56% of patients. All of the tumour responses were observed in patients with ovarian cancer, for an overall response rate of 23% in this subgroup. Of the evaluable patients with ovarian cancer, nine (35%) had been treated with three or more prior lines of therapy.
Discussion
The recent availability of an increasing number of promising investigational and newly approved drugs has rechallenged the role of multiagent chemotherapy in the clinical management of solid tumours with greater impetus for rational combinations of chemotherapy that maximise efficacy with minimal toxicity (Cannistra, 2002). However, the best approach for combining topotecan with other anticancer agents has been debated because additive myelosuppresssion can markedly limit the doses of drugs that can be safely administered together (Cannistra, 1999). Etoposide and topotecan are both commonly used agents with potential synergy. This study demonstrates that oral etoposide 35 mg m−2 b.i.d. D1–5 and 1.8 mg m−2 96 h (total dose) infusional topotecan can be administered on an alternating continual weekly schedule for at least 12 weeks, with promising clinical activity. Furthermore, this potentially maximises the benefit of alternating induction of topoisomerase I and II. The regimen was found to be a well tolerated, although a relatively inconvenient, outpatient treatment. Toxicity is characterised by neutropenia, and thrombocytopenia was dose-limiting with nonhaematological toxicity being notably mild.
DNA topoisomerases I and IIa are targets for many clinically important antineoplastic agents. The drugs that interact with topoisomerase I, the camptothecins, are structurally distinct from the topoisomerase II-interacting agents of the anthracycline, epipodophyllotoxin and anthracenedione classes. No clinically useful agent that preferentially targets topoisomerase IIβ has been developed to date. Maximising the utility of topoisomerase-interacting agents, by sequential combination, may also minimise the development of resistance (Potmesil et al, 1988; Sugimoto et al, 1990). Although p-glycoprotein overexpression is likely the most important, other mechanisms probably also contribute, and continual, rather than intermittent exposure may limit the development of resistance (Schneider et al, 1994). Continual exposure will also limit the development of resistance. However, whether significant benefit comes from a greater total dose, longer exposure of a greater number of cycling cells or time above a concentration threshold is unclear (Joel, 1996).
The rationale for choice of DLs appears to have been justified by the results. Protracted oral etoposide dose in combination treatment is typically dosed at 50–75 mg m−2 day−1 (Hainsworth, 1999). There is limited data on topotecan by protracted infusion in combination regimens. Hochster et al reported an MTD of 0.4 mg m−2 day−1 D1–14 q 21 in combination with short infusion paclitaxel (Chachoua et al, 1997) and we defined an MTD for topotecan of 0.7 mg m−2 day−1 by 96 h infusion in combination with 96 h infusion paclitaxel (Penson et al, 2001). Doxorubicin and topotecan administered sequentially could be combined at 60–70% of the typical dose (Seiden et al, 2002). With the protocol design calling for protracted sequential treatment without a pause to allow for recovery of bone marrow function, the starting doses were reduced a further 50%. The dose reduction strategy set stringent thresholds for continued treatment in an attempt to prevent severe myelosuppression. Given the variable and unpredictable bioavailability of oral etoposide, and the resulting variable tolerability, there was surprisingly little intrapatient variability in haematological and nonhaematological toxicity during the study. However, on three occasions, the DL cohorts had to be modified to accrue more patients.
Topotecan appears to be a schedule-dependent cytotoxic (Rowinsky et al, 1992). Phase I and II clinical trials have been performed to evaluate the administration of topotecan by continuous i.v. infusion for periods of 24 h (van Warmerdam et al, 1995; Abbruzzese et al, 1996; Hoskins et al, 1998), 72 h (Burris et al, 1994), 120 h (Burris et al, 1994) and 21 days (Hochster et al, 1994). The Canadian National Cancer Institute conducted a randomised phase II trial to investigate the schedule dependence of topotecan (Hoskins et al, 1998). Patients received topotecan given according to the approved dosing regimen, 1.5 mg m−2 i.v. over 30 min on 5 consecutive days every 21 days, or by 24-h i.v. infusion of a 1.75 mg m−2 dose once a week for 4 weeks, with cycles repeated every 6 weeks. The results revealed that the response rate for topotecan given by the D1–5 dosing regimen was significantly better than the 24-h infusion schedule (22.6 vs 3.1%, P=0.026). The weekly 24-h infusion schedule was clearly associated with less severe haematological toxicity than the approved dosing regimen. However, the investigators did not believe that the dose, which had been previously defined in a phase I study (Haas et al, 1994), could be significantly increased. In contrast, there are more encouraging results from studies of topotecan delivered by protracted infusion, in combination with other chemotherapeutic agents (Hochster et al, 1994), and our own experience with 96 h infusion topotecan has been very positive (Penson et al, 2001).
While the hope has been to exploit a therapeutic window, combining topoisomerase I and II inhibitors has been associated with substantial toxicity. Studies have, in fact, not infrequently defined an MTD lower than the initial DL (Karato et al, 1993; Ando et al, 1997). The commonest combinations have been irinotecan given concurrently with etoposide (Karato et al, 1993; Masuda et al, 1994; Oshita et al, 1997; Masuda et al, 1998), in which the DLTs were typically neutropenia and diarrhoea, and required G-CSF support. A study of sequential irinotecan followed by etoposide reported a similar MTD (Ando et al, 1997). Other sequential studies have investigated topotecan and etoposide (Herben et al, 1997; Hammond et al, 1998) or doxorubicin and topotecan (Tolcher et al, 1997; Seiden et al, 2002). This study reports a particularly favourable toxicity profile, with encouraging efficacy. Although oral etoposide potentially offers a superior schedule of dosing (Slevin et al, 1989), this has been questioned (Girling, 1996). Limited data is available about oral topotecan. However, oral topotecan, or an alternate bioavailable camptothecin analogue, would obviate the logistical challenges of infusional chemotherapy and this option is an obvious avenue of investigation and is being discussed.
In a heavily pretreated population of patients, the response rate was impressive. There were six radiologically confirmed responses (18%), three of which were confirmed complete radiological remissions. The majority (56%) of patients had a tumour response or stable disease, typically tolerated with minimally toxic treatment. Although little more than a proof of principle, the approach of delivering minimally toxic treatment to ‘hold the disease at bay’, or ‘shrink’ the disease may have delivered blood concentrations above the therapeutic threshold but below those associated with significant toxicity. However, this therapeutic window is far from defined, and is complex given the attempt to concurrently escalate both drugs.
In summary, this study has demonstrated that oral etoposide 35 mg m−2 b.i.d. D1–5 and 1.8 mg m−2 96 h (total dose) infusional topotecan can be administered on an alternating continual weekly schedule for at least 12 weeks with promising clinical activity that merits a phase II study in patients with ovarian cancer.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Abbruzzese JL, Madden T, Sugarman SM, Ellis AL, Loughlin S, Hess KR, Newman RA, Zwelling LA, Raber MN (1996) Phase I clinical and plasma and cellular pharmacological study of topotecan without and with granulocyte colony-stimulating factor. Clin Cancer Res 2: 1489–1497
Ando M, Eguchi K, Shinkai T, Tamura T, Ohe Y, Yamamoto N, Kurata T, Kasai T, Ohmatsu H, Kubota K, Sekine I, Hojo N, Matsumoto T, Kodama T, Kakinuma R, Nishiwaki Y, Saijo N (1997) Phase I study of sequentially administered topoisomerase I inhibitor (irinotecan) and topoisomerase II inhibitor (etoposide) for metastatic non-small-cell lung cancer. Br J Cancer 76: 1494–1499
Burris III HA, Awada A, Kuhn JG, Eckardt JR, Cobb PW, Rinaldi DA, Fields S, Smith L, Von Hoff DD (1994) Phase I and pharmacokinetic studies of topotecan administered as a 72 or 120 h continuous infusion. Anticancer Drugs 5: 394–402
Cannistra SA (1999) Back to the future: multiagent chemotherapy in ovarian cancer revisited. J Clin Oncol 17: 741–743
Cannistra SA (2002) Is there a ‘best’ choice of second-line agent in the treatment of recurrent, potentially platinum-sensitive ovarian cancer? J Clin Oncol 20: 1158–1160
Caron P, Wang J (1993) DNA topoisomerases as a target of therapeutics: a structural overview. In Molecular Biology of DNA Topoisomerases and its Application to Chemotherapy Andoh T, Ikeda H, Oguro M (eds) pp 1–18. Boca Ratan, FL: CRC Press, Inc
Chachoua A, Hochster H, Sorich J, Wasserheit C, Taubes B, Friedberg A, Arbuck A, Speyer J (1997) Phase I study of paclitaxel (P) combined with 14-day topotecan (T) continuous IV (CIV) infusion in previously treated and untreated patients (pts). Proc ASCO; abstract 839
Collins JM, Grieshaber CK, Chabner BA (1990) Pharmacologically guided phase I clinical trials based upon preclinical drug development. J Natl Cancer Inst 82: 1321–1326
Eder JP, Chan V, Wong J, Wong YW, Ara G, Northey D, Rizvi N, Teicher BA (1998) Sequence effect of irinotecan (CPT-11) and topoisomerase II inhibitors in vivo. Cancer Chemother Pharmacol 42: 327–335
Furman L, Camitta BM, Jaffe N, Sallan SE, Cassady JR, Traggis D, Leavitt P, Nathan DG, Frei III E (1976) Development of an effective treatment program for childhood acute lymphocytic leukemia: a preliminary report. Med Pediatr Oncol 2: 157–166
Girling DJ (1996) Comparison of oral etoposide and standard intravenous multidrug chemotherapy for small-cell lung cancer: a stopped multicentre randomised trial. Medical Research Council Lung Cancer Working Party. Lancet 348: 563–566
Gupta M, Abdel-Megeed M, Hoki Y, Kohlhagen G, Paull K, Pommier Y (1995) Eukaryotic DNA topoisomerases mediated DNA cleavage induced by a new inhibitor: NSC 665517. Mol Pharmacol 48: 658–665
Haas NB, LaCreta FP, Walczak J, Hudes GR, Brennan JM, Ozols RF, O'Dwyer PJ (1994) Phase I/pharmacokinetic study of topotecan by 24-hour continuous infusion weekly. Cancer Res 54: 1220–1226
Hainsworth JD (1999) Extended-schedule oral etoposide in selected neoplasms and overview of administration and scheduling issues. Drugs 58: 51–56
Hammond LA, Eckardt JR, Ganapathi R, Burris HA, Rodriguez GA, Eckhardt SG, Rothenberg ML, Weiss GR, Kuhn JG, Hodges S, Von Hoff DD, Rowinsky EK (1998) A phase I and translational study of sequential administration of the topoisomerase I and II inhibitors topotecan and etoposide. Clin Cancer Res 4: 1459–1467
Heck MM, Earnshaw WC (1986) Topoisomerase II: a specific marker for cell proliferation. J Cell Biol 103: 2569–2581
Herben VM, ten Bokkel Huinink WW, Dubbelman AC, Mandjes IA, Groot Y, van Gortel-van Zomeren DM, Beijnen JH (1997) Phase I and pharmacological study of sequential intravenous topotecan and oral etoposide. Br J Cancer 76: 1500–1508
Hochster H, Liebes L, Speyer J, Sorich J, Taubes B, Oratz R, Wernz J, Chachoua A, Raphael B, Vinci RZ (1994) Phase I trial of low-dose continuous topotecan infusion in patients with cancer: an active and well-tolerated regimen. J Clin Oncol 12: 553–559
Holm C, Goto T, Wang JC, Botstein D (1985) DNA topoisomerase II is required at the time of mitosis in yeast. Cell 41: 553–563
Hoskins P, Eisenhauer E, Beare S, Roy M, Drouin P, Stuart G, Bryson P, Grimshaw R, Capstick V, Zee B (1998) Randomized phase II study of two schedules of topotecan in previously treated patients with ovarian cancer: a National Cancer Institute of Canada Clinical Trials Group study. J Clin Oncol 16: 2233–2237
Joel S (1996) The clinical pharmacology of etoposide: an update. Cancer Treat Rev 22: 179–221
Karato A, Sasaki Y, Shinkai T, Eguchi K, Tamura T, Ohe Y, Oshita F, Nishio M, Kunikane H, Arioka H (1993) Phase I study of CPT-11 and etoposide in patients with refractory solid tumors. J Clin Oncol 11: 2030–2035
Kaufmann SH (1991) Antagonism between camptothecin and topoisomerase II-directed chemotherapeutic agents in a human leukemia cell line. Cancer Res 51: 1129–1136
Kohn KW, Pommier Y (2000) Molecular and biological determinants of the cytotoxic actions of camptothecins. Perspective for the development of new topoisomerase I inhibitors. Ann N Y Acad Sci 922: 11–26
Liebes L, Potmesil M, Kim T, Pease D, Buckley M, Fry D, Cho J, Adler H, Dar K, Zeleniuch-Jacquotte A, Hochster H (1998) Pharmacodynamics of topoisomerase I inhibition: Western blot determination of topoisomerase I and cleavable complex in patients with upper gastrointestinal malignancies treated with topotecan. Clin Cancer Res 4: 545–557
Liu LF (1989) DNA topoisomerase poisons as antitumor drugs. Annu Rev Biochem 58: 351–375
Masuda N, Fukuoka M, Kudoh S, Matsui K, Kusunoki Y, Takada M, Nakagawa K, Hirashima T, Tsukada H, Yana T (1994) Phase I and pharmacologic study of irinotecan and etoposide with recombinant human granulocyte colony-stimulating factor support for advanced lung cancer. J Clin Oncol 12: 1833–1841
Masuda N, Matsui K, Negoro S, Takifuji N, Takeda K, Yana T, Kobayashi M, Hirashima T, Kusunoki Y, Ushijima S, Kawase I, Tada T, Sawaguchi H, Fukuoka M (1998) Combination of irinotecan and etoposide for treatment of refractory or relapsed small-cell lung cancer. J Clin Oncol 16: 3329–3334
Oshita F, Noda K, Nishiwaki Y, Fujita A, Kurita Y, Nakabayashi T, Tobise K, Abe S, Suzuki S, Hayashi I, Kawakami Y, Matsuda T, Tsuchiya S, Takahashi S, Tamura T, Saijo N (1997) Phase II study of irinotecan and etoposide in patients with metastatic non-small-cell lung cancer. J Clin Oncol 15: 304–309
Penson RT, Supko JG, Seiden MV, Fuller AF, Berkowitz RS, Goodman A, Campos SM, MacNeill KM, Cook S, Matulonis UA (2001) A Phase I–II study of 96-hour infusional topotecan and paclitaxel for patients with recurrent Mullerian tumors. Cancer 92: 1156–1167
Potmesil M, Hsiang YH, Liu LF, Bank B, Grossberg H, Kirschenbaum S, Forlenza TJ, Penziner A, Kanganis D, Forlenzar TJ (1988) Resistance of human leukemic and normal lymphocytes to drug-induced DNA cleavage and low levels of DNA topoisomerase II. Cancer Res 48: 3537–3543
Rowinsky EK, Grochow LB, Hendricks CB, Ettinger DS, Forastiere AA, Hurowitz LA, McGuire WP, Sartorius SE, Lubejko BG, Kaufmann SH (1992) Phase I and pharmacologic study of topotecan: a novel topoisomerase I inhibitor. J Clin Oncol 10: 647–656
Schneider E, Horton JK, Yang CH, Nakagawa M, Cowan KH (1994) Multidrug resistance-associated protein gene overexpression and reduced drug sensitivity of topoisomerase II in a human breast carcinoma MCF7 cell line selected for etoposide resistance. Cancer Res 54: 152–158
Seiden MV, Ng SW, Supko JG, Ryan DP, Clark JW, Lynch T, Huang KC, Kwiatkowski D, Skarin A, Eder Jr JP (2002) A phase I clinical trial of sequentially administered doxorubicin and topotecan in refractory solid tumors. Clin Cancer Res 8: 691–697
Slevin ML, Clark PI, Joel SP, Malik S, Osborne RJ, Gregory WM, Lowe DG, Reznek RH, Wrigley PF (1989) A randomized trial to evaluate the effect of schedule on the activity of etoposide in small-cell lung cancer. J Clin Oncol 7: 1333–1340
Sugimoto Y, Tsukahara S, Oh-hara T, Isoe T, Tsuruo T (1990) Decreased expression of DNA topoisomerase I in camptothecin-resistant tumor cell lines as determined by a monoclonal antibody. Cancer Res 50: 6925–6930
Tolcher AW, O'Shaughnessy JA, Weiss RB, Zujewski J, Myhand RC, Schneider E, Hakim F, Gress R, Goldspiel B, Noone MH, Brewster LR, Gossard MR, Cowan KH (1997) A phase I study of topotecan followed sequentially by doxorubicin in patients with advanced malignancies. Clin Cancer Res 3: 755–760
van Warmerdam LJ, ten Bokkel Huinink WW, Rodenhuis S, Koier I, Davies BE, Rosing H, Maes RA, Beijnen JH (1995) Phase I clinical and pharmacokinetic study of topotecan administered by a 24-hour continuous infusion. J Clin Oncol 13: 1768–1776
Acknowledgements
We thank Moira Kirvan, Eileen Regan, Aimee Harris, Ann Willman, Tina Colella, Eleanor Chung, Barbara Ratner, Ilya Dixon and Marietta Palacay-Radona for their excellent assistance in the conducting of this study. Emil Frei III, MD is rightly credited as the father of the strategy of continual treatment in the hope of improving the efficacy of schedule-dependent cytotoxic chemotherapy and we acknowledge his outstanding contribution to the field. This study is supported in part by a grant from Smith Kline Beecham (King of Prussia, PA).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Penson, R., Seiden, M., Matulonis, U. et al. A phase I clinical trial of continual alternating etoposide and topotecan in refractory solid tumours. Br J Cancer 93, 54–59 (2005). https://doi.org/10.1038/sj.bjc.6602671
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6602671
