
Abstract
Angiogenesis is crucial for tumour growth and the formation of metastases. Various classes of angiogenesis inhibitors that are each able to inhibit one of the various steps of this complex process can be distinguished. Results from clinical studies with these agents are summarised. In general, it has been shown that most angiogenesis inhibitors can be safely administered, but that tumour regressions are rare. Combining angiogenesis inhibitors with cytotoxic chemotherapy can enhance anticancer activity. Recently, some promising data with regard to clinical efficacy have been presented. While performing clinical studies with angiogenesis inhibitors, defining biological activity is crucial, but thus far no validated techniques are available. It is conceivable that in the near future various classes of angiogenesis inhibitors will be combined in an attempt to further improve antiangiogenic and anticancer activity.
Similar content being viewed by others

Main
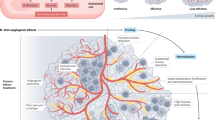
In order to grow and metastasise, tumours exceeding the size of 1 mm3 are dependent on blood supply from newly formed adjacent blood vessels.
Tumour-related angiogenesis is a multistep process that is initiated through the activity of various proangiogenic factors or stimuli that are secreted by tumour cells and host components such as macrophages, lymphocytes and kidney cells (Klagsbrun and D' Amore, 1996). Of these proangiogenic factors, vascular endothelial growth factor (VEGF) is predominant. Vascular endothelial growth factor binds with high affinity to the transmembrane endothelial VEGF receptor type 1 and 2 (VEGFR-1 or flt-1 and VEGFR-2 or FLK-1/KDR, respectively). Ligand–receptor interaction initiates a cascade of intracellular signals through increased receptor tyrosine kinase activity, resulting in endothelial cell proliferation and the formation of new blood vessels. Increased expression of VEGF is associated with poor clinical outcome irrespective of tumour stage or tumour grade.
Besides VEGF, basic fibroblast growth factor (bFGF), platelet-derived growth factor (PDGF), interleukin-8 and insulin-like growth factor (IGF) are factors involved in angiogenesis, and these factors have their own endothelial cell receptors.
As angiogenesis is of crucial importance for tumour growth, inhibiting this process has become a major challenge in the development of new anticancer agents.
Tumour-related angiogenesis is dependent on specific growth factors, the presence and activation of endothelial cell receptors, endothelial cell proliferative activity as such, and the interaction between proliferating endothelial cells and extracellular matrix components. All these specific steps can be inhibited by targeted agents.
Neutralising VEGF prior to its attachment to endothelial receptors can be accomplished by monoclonal antibodies. Inhibiting the tyrosine kinase activity of VEGFR-2, PDGF and/or bFGF receptors following ligand binding can be achieved through specific inhibitors. Inhibiting endothelial cell proliferation can be achieved by mimicking the activity of naturally occurring antiangiogenic proteins such as thrombospondin-1 (TSP-1), endostatin and angiostatin. Inhibiting the interaction between proliferating endothelial cells and various extracellular matrix components can be accomplished by inhibiting the activity of the transmembrane endothelial cell integrins αvβ3 and αvβ5. In contrast to quiescent blood vessels where expression of integins is minimal, actively proliferating endothelial cells express integrins to a high extent. Integrin activity can be inhibited by antibodies or small molecule inhibitors.
A different approach that may lead to diminished tumour vascularisation is the destruction of established vasculature. The so-called ligand-directed vascular targeting agents (VTAs) are able to induce a rapid and specific shutdown of tumour blood supply either by using antibodies, peptides or growth factors that deliver toxins and procoagulants or apoptotic effectors specifically to the tumour endothelium. The so-called small-molecule VTAs try to recognise the pathophysiological differences between the normal and tumour endothelium, and are thus able to induce vascular shutdown, specifically in tumours.
In this review, we will give an overview of the results of clinical studies that have been performed with various classes of antiangiogenic and antivascular agents (Table 1). While discussing the results of these studies, we will focus in more detail on the mechanisms of action, and we will discuss some of the difficulties that have been faced while performing these studies. Finally, we will give some thoughts on where to go in future studies.
Where are we now?
Monoclonal antibodies directed against VEGF
Bevacizumab is a humanised monoclonal VEGF antibody with antiangiogenic and antitumour activity in preclinical models. Phase I studies exploring different intravenous (i.v.) administration schedules showed that the agent, given as single agent or in combination with various cytotoxic agents, was safe and did not cause dose-limiting toxicity (DLT) when given at predefined doses. Some bleeding episodes were seen that were not attributed to the agent. Antiangiogenic activity was assessed by contrast-enhanced dynamic magnetic resonance imaging (DEMRI), showing a decrease in tumour microvascular permeability. Biological activity was also demonstrated by a decline in serum levels of unbound VEGF (Gordon et al, 2001; Margolin et al, 2001; Langmuir et al, 2002).
Phase II studies in patients with various tumour types showed partial tumour regressions and/or increased time to progression (compared to placebo or cytotoxic treatment alone), when bevacizumab was given in various doses and different dosing intervals, either as single agent or in combination with various cytotoxic agents. In these studies, the increased incidence of thromboembolic complications, bleeding episodes (some of which were lethal), hypertension and proteinuria was the reason for concern (Burnstein et al, 2002; Miller et al, 2002; Yang et al, 2002; Kabbinavar et al, 2003). Two randomised phase III studies combining bevacizumab with standard cytotoxic treatment in patients with metastatic breast cancer showed disappointing results (Miller et al, 2002,2003), but a recently presented randomised phase III study in 800 previously untreated patients with metastatic colorectal cancer comparing treatment with irinotecan, 5-fluorouracil and leucovorin (IFL) and IFL combined with bevacizumab given every 2 weeks showed increased response rates, prolonged time to progression and significantly increased survival with no increased incidence of thromboembolic complications (Hurwitz et al, 2003).
VEGF-trap is a specific antagonist that binds to and subsequently inactivates VEGF. It is claimed to have a higher affinity for VEGF than monoclonal antibodies, and can be administered subcutaneously. Phase I studies are ongoing (Dupont et al, 2003).
VEGFR-2 antibodies
As ligand–receptor interaction of VEGF and VEGFR-2 is crucial for endothelial proliferation, it is conceivable that apart from blocking VEGF as the principal ligand, blocking VEGFR-2 is another rational approach to prevent this cascade. However, monoclonal antibodies targeting VEGFR-2 have not yet been tested extensively and, to date, only one phase I study with such a compound, IMC-1C11, has been published. No major drug-related toxicity was seen in this small study (Posey et al, 2003).
VEGF receptor tyrosine kinase inhibitors
SU5416 showed antiangiogenic and antitumour activity in preclinical studies, and was the first VEGF receptor tyrosine kinase inhibitor to be tested clinically (Stopeck et al, 2003).
Phase I trials yielded headache and vomiting as DLT. Due to a short half-life, SU5416 had to be given frequently in order to achieve active plasma concentrations for prolonged periods of time. The twice-weekly treatment schedule was studied most often, but even this schedule yielded active plasma concentrations for only a limited period of time. A more frequent intravenous administration schedule was considered to be unpractical, and limited bioavailability hampered the development of an oral compound. Although some clinical activity was recorded, and DEMRI analysis revealed a reduction in tumour vascular permeability, biological activity could not be demonstrated by means of consistent changes in the serum markers of endothelial cell proliferation. In phase I and II combination trials, SU5416 could be given safely in combination with 5-FU and leucovorin, but combining SU5416 with cisplatinum and gemcitabine yielded an unexpectedly high number of severe thromboembolic complications, leading to premature closing of the study (Kuenen et al, 2002a). Randomised phase III studies of SU5416 with 5-FU/leucovorin and 5-FU/leucovorin/irinotecan in patients with metastatic colorectal carcinoma failed to show a survival benefit of the SU5416 containing regimens, resulting in the cessation of further development of this compound.
SU 6668 is an orally available inhibitor of VEGF, bFGF and PDGF receptor tyrosine kinase activity. Phase I trials showed good tolerability with once daily dosing, but a twice daily administration schedule yielded DLT consisting of fatigue, dyspnea, chest pain and pericardial effusions (Rosen et al, 2001; Brahmer et al, 2002). Pharmacokinetic analysis revealed a short plasma half-life, with plasma concentrations exceeding those that inhibited tyrosine kinase activity in preclinical models in the first day, but not following prolonged administration. Due to these disappointing results, and despite the fact that some clinical activity was noted in clinical studies, the compound was withdrawn from further development (Britten et al, 2002; Kuenen et al, 2002b; Ueda et al, 2002).
SU11248 is a broad-spectrum orally available tyrosine kinase inhibitor, inhibiting VEGF, PDGF, c-Kit and Flt-3 kinase activity (Mendel et al, 2002). This inhibitory spectrum makes this agent to be more than a specific antiangiogenic agent. The recently presented phase I trials exploring either 14 or 28 days treatment followed by 14 days off treatment yielded DLT consisting of fatigue, lethargy and myelosuppression. Two patients died due to drug-related neutropenic sepsis. Tumour regressions have been noted, and positron emission tomography (PET) imaging studies and a rise in serum VEGF levels parallelled by a consistent decrease in soluble VEGFR-2 levels indicate antiangiogenic activity (Manning et al, 2003; Raymond et al, 2003; Rosen et al, 2003; Toner et al, 2003). Phase II trials are being initiated.
PTK787/ZK22854 is an orally available VEGFR-1 and VEGFR-2 tyrosine kinase inhibitor. Ataxia, vertigo and hypertension were noted as DLTs, whereas some cases of venous thromboembolism were recorded (Yung et al, 2002). Biological activity has been demonstrated as DEMRI analysis showed a dose-dependent decrease in blood flow in hepatic metastases of colorectal cancer patients (Thomas et al, 2001; Drevs et al, 2002; Yung et al, 2003). Phase II and III trials with PTK in combination with 5-fluorouracil, irinotecan and/or oxaliplatin in patients with metastatic colorectal cancer have been performed or are currently ongoing (Steward et al, 2003; Trarbach et al, 2003).
ZD6474 inhibits both VEGF and epidermal growth factor (EGF) receptor tyrosine kinase activity. Following prolonged continuous oral administration, DLT consisted of diarrhoea, hypertension, hepatic toxicity and cutaneous rash, whereas asymptomatic QTc prolongation was seen throughout the dose levels studied (Hurwitz et al, 2002; Minami et al, 2003). Hints of clinical activity were recorded in some patients. It is conceivable that diarrhoea and skin rash are due to the inhibition of the EGF receptor tyrosine kinase activity, illustrating the broad range of tyrosine kinase inhibition by this agent.
CP-547,632 is a selective VEGFR-2 receptor tyrosine kinase inhibitor that is currently in phase I studies (Tolcher et al, 2002).
Naturally occurring inhibitors of endothelial cell proliferation
Thrombospondin-1 is a naturally occurring inhibitor of endothelial cell proliferation. As TSP-1 is a large protein, restricting its pharmacological use, and as its antiangiogenic effects are restricted to the N-terminal region, various structural modifications have been made, leading to TSP-1 mimetic proteins. ABT-510 is such a protein that can be administered subcutaneously. Phase I studies have shown excellent tolerability at all dose levels tested, also following prolonged administration (De Vos et al, 2002; Gordon et al, 2003). Pharmacokinetic analysis has shown that plasma concentrations exceeding those inhibiting angiogenesis in preclinical studies could be maintained for prolonged periods of time following once or twice daily administration. Phase I combination studies are ongoing, whereas single-agent phase II studies in NSCLC and renal carcinoma have been initiated.
Endostatin and angiostatin are both naturally occurring angiogenesis inhibitors that are able to induce the apoptosis of endothelial cells and to inhibit endothelial cell migration and proliferation. Endostatin is a 20 kDa fragment derived from the C-terminal region of collagen XVIII, angiostatin is a 38 kDa fragment of plasminogen. Various mechanisms of action are considered for these antiangiogenic properties.
Phase I studies with recombinant human endostatin given as daily i.v. bolus injections revealed no drug-related toxicity and some hints of clinical activity. The pharmacokinetic analyses showed that, at the highest dose that could be practically administered, drug exposure was lower than that yielding maximum tumour growth inhibition in preclinical studies. Additionally, DEMRI analysis done in a number of patients did not show any relevant changes in perfusion throughout the treatment period (Eder et al, 2002) and analysis of various serum markers of angiogenesis was inconclusive (Herbst et al, 2002). Currently, studies exploring continuous intravenous and subcutaneous administration of endostatin are being performed, with preliminary data showing increased drug exposure when compared to bolus administration (Hansma et al, 2002; Tran et al, 2002).
Angiostatin is currently undergoing phase I clinical trials, exploring both i.v. and twice daily subcutaneous injections (Beerepoot et al, 2001; DeMoraes et al, 2001; Voest et al, 2002).
Other inhibitors of endothelial cell proliferation
TNP-470 is a derivative of the natural compound fumagilin that showed antiangiogenic activity in preclinical models through inhibition of cyclin-dependent kinase 2, retinoblastoma protein phosphorylation and methionine aminopeptidase.
The first clinical studies with TNP-470 explored weekly 1-h infusions, but as the terminal plasma half-life turned out to be only minutes, additional studies with either more frequent administrations or more protracted infusion schedules (up 120 h) were performed. Dose-limiting toxicity consisted of neuropsychiatric effects (Bhargava et al, 1999; Stadler et al, 1999; Logothetis et al, 2001). More recently, TNP-470 administered as a protracted infusion in combination with paclitaxel and carboplatin has yielded interesting efficacy data in patients with NSCLC (Blumenschein et al, 2002).
Although the exact mechanism of antiangiogenic action of thalidomide has not been fully elucidated, a large number of clinical studies with this compound have been performed. In particular, patients with multiple myeloma seem to benefit from thalidomide, although some responses have also been recorded in solid tumours. Obstipation, lethargy and peripheral neuropathy have been ascribed to thalidomide.
Vitaxin is a monoclonal antibody of the integrin αvβ3 that can be administered subcutaneously. A phase I trial showed good clinical tolerability and antitumour activity in a patient with leiomyosarcoma. Direct antiangiogenic activity was not demonstrated (Gutheil et al, 2000). A phase II study in patients with leiomyosarcomas did not confirm antitumour activity (Patel et al, 2001).
Medi-522 is a novel humanised monoclonal antibody targeting the integrin αvβ3 receptor that is currently undergoing phase I studies (Faivre et al, 2003).
Cilengitide is a small molecule inhibitor of the integrins αvβ3 and αvβ5. A phase I study exploring a continuous twice weekly i.v. administration schedule showed excellent clinical tolerability with no drug-related side effects following a predefined dose escalation schedule (Eskens et al, 2003). Pharmacokinetic analysis demonstrated that plasma concentrations exceeding those that yielded optimal antiangiogenic activity in preclinical models were reached. Currently, aditional pharmacodynamic analysis to explore biological activity is being done (Holden et al, 2002).
Vascular targeting agents
Combretastatin prodrug A4 (CA4P) is a tubulin-depolymerising VTA that is able to induce a rapid change in endothelial cell shape, causing a disruption of the endothelial layer, vascular congestion and loss of blood flow. Preclinical studies showed selectivity for rapidly proliferating tumour endothelium, leaving mature endothelial cells unaffected. Dose-limiting toxicity in three phase I studies, exploring different treatment schedules, consisted of acute coronary syndrome, tumour pain and ataxia. QTc prolongation, cutaneous flushing and hot flashes and diffuse abdominal pain and tumour pain were associated with the drug infusion. One pathological complete response lasting 33 months was recorded in a patient with an anaplastic thyroid carcinoma.
Reversible decreases of tumour blood flow were observed using either dynamic enhanced MRI scanning or PET scanning (Stevenson et al, 2000; Rustin et al, 2001; Dowlati et al, 2002).
AVE8062A is a combretastatin analogue that is undergoing phase I testing. Transient and reversible myocardial and central nervous system ischaemia were recorded at the highest dose levels studied (Tolcher et al, 2003).
ZD6126 binds to endothelial tubulin, and is able to induce changes in endothelial shape, leading to vascular shutdown. Preclinical studies showed that only immature endothelial cells responded to ZD6126, whereas mature endothelial cells were not affected.
In a phase I study exploring a 10-min single i.v. administration once every 21 days, abdominal pain and gastrointestinal toxicity were dose limiting. Significant changes in tumour blood flow were observed, as some cases of prolonged stable disease (DelProposto et al, 2002; Gadgeel et al, 2002). A second phase I study is exploring a weekly administration schedule (Radema et al, 2002).
DMXAA is a flavonoid that, in preclinical models, is able to induce TNF-α production and induce vascular disruption, selectively in the tumour microenvironment.
In a phase I trial, DLT consisted of urinary incontinence, various seemingly central nervous system-related toxicities, dyspnea and cardiac toxicity (Rustin et al, 2003).
Biological activity was assessed by means of DEMRI analysis, showing changes in tissue perfusion, vessel permeability and vessel surface area (Galbraith et al, 2002).
Where are we going?
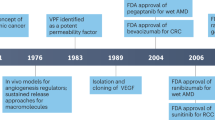
The first clinical studies with antiangiogenic agents were initiated 5 years ago, when preclinical studies in mice showed tumour disappearance and prolonged survival following the administration of endostatin and angiostatin. Scientists and especially the lay press were excited over these results.
Clinical studies of these 5 years have shown that many angiogenesis inhibitors can be given safely to patients. Some antiangiogenic agents were even completely devoid of toxicity, and defining an optimal dose for subsequent activity testing of these agents was difficult, if not impossible. In the meantime, it became obvious that antiangiogenic activity of some agents occurred at very low doses, even more questioning the concept of dose escalation until toxicity. Determining optimal biological activity became an important new end point in phase I studies.
How can we optimally define the optimal biologically effective dose, and which pharmacodynamic analysis should be used for this?
Target inhibition, dormancy of endothelial cells or vascular shutdown with subsequent tumour ischaemia are important biologial end points that have the potential to predict antitumour activity, but the only reliable source to determine these end points is the tumour. Here we are facing practical problems, because most physicians consider taking repeated tumour biopsies to be very cumbersome for patients. Although some authors consider taking these biopsies repeatedly to be feasible (Dowlati et al, 2001), the number of studies incorporating these procedures is still small. Although many alternative procedures and strategies have been developed to circumvent this problem, it is conceivable that, in future studies, exploring antiangiogenic agents taking tumour biopsies will become mandatory.
While trying to demonstrate biological activity in a less invasive manner, various surrogate markers of antiangiogenic or antivascular activity have been studied. The analysis of serum markers of endothelial cell proliferation thus far has failed to show consistent changes following treatment with most antiangiogenic agents, but changes in serum concentrations of VEGF and soluble VEGFR-2 seem to have more potential to predict tumour hypoxia (Drevs et al, 2003; Toner et al, 2003). The pathophysiological mechanisms that explain these changes are still a matter of debate, however, as tumour hypoxia as such has not been demonstrated in human tumours.
Noninvasive techniques such as DEMRI and PET scans are increasingly used to demonstrate the biological activity of antiangiogenic agents or VTAs. These techniques can show changes in tumour blood flow and tumour viability, respectively. In numerous studies, DEMRI and PET scan studies were able to demonstrate these pharmacodynamic effects, but it must be realised that both techniques are still unvalidated to predict antitumour activity or patient benefit. Only large clinical studies correlating the outcomes of DEMRI and PET scan studies with clinical outcome can definitely assess the role of these techniques in future patient management.
Another method to assess the biological activity of antiangiogenic agents is by the use of pharmacokinetic studies. Drug exposure or peak plasma concentrations can be used to determine the dose that is most likely to give antiangiogenic or antitumour activity, but here it has to be realised that comparing human plasma concentrations to target inhibitory concentrations in preclinical models cannot and will not always be predictive for the same biological effect. Nevertheless, pharmacokinetic analysis will remain of crucial importance to prevent unnecessary dose escalation in case of incomplete or saturated absorption with maximal drug exposure, and to show the relations between clinical toxicity and plasma concentrations.
Antiangiogenic agents are cytostatic agents that are not likely to cause tumour regressions in patients with advanced or metastatic cancer. In order to assess their antitumour activity in phase II studies, combining these agents with cytotoxic anticancer agents seems to be a logical step. Clinically, the combination of two antiangiogenic agents, each having a distinctive inhibitory mechanism of action, has not been tested thus far. A large number of studies combining cytotoxic and antiangiogenic agents have been performed. These phase II studies most often had a randomised design, comparing the response rates or time to disease progression. In order to define safe combination dosages, these studies were always preceded by phase I combination studies showing the safety of these combinations.
As some of these randomised phase II trials were indeed able to demonstrate improved antitumour activity of the combination treatment, results of large randomised phase III trials were eagerly awaited. However, until recently, these phase III trials, mostly done in patients with metastatic colorectal or breast cancer, all failed to show improved survival, and serious concern was growing as to whether we would ever be able to demonstrate the clinical efficacy of any of these exciting and promising agents.
It was with great excitement, therefore, that recently the first randomised phase III trial in patients with metastatic colorectal cancer showed improved survival when cytotoxic treatment was combined with bevacizumab. It is this trial that, on the one hand, has reconfirmed the conviction that we, currently, are testing effective new agents, whereas, on the other hand, it has become clear that the only way to assess clinical benefit or a complete generation of these agents will be through these large, costly and time-consuming randomised trials. Besides, we have to accept that more than one randomised trial will be needed, as at this moment it is not even fully clear that the dose tested in this study is in fact the most optimal one. Another intriguing question that remains is why the addition of bevacizumab was beneficial to this group of colorectal carcinoma patients, whereas results of randomised studies with various other antiangiogenic agents in a comparable population of patients all turned out to be negative.
If one is to consider the currently perceived limited antitumour activity of single-agent antiangiogenic treatment, it can be argued that a combination of antiangiogenic agents with different targets or a combination of antiangiogenic agents and VTAs might show increased activity. Preclinical studies exploring such combinations are now being performed, and some promising data have been presented. In addition to this, it is also conceivable that combining antiangiogenic agents with other targeted agents should be explored, but for this the help of pharmaceutical companies who are willing to share their compounds and knowledge is urgently needed.
If one is still to explore single agent antiangiogenic treatment, a situation of minimal residual disease following either cytoreductive surgery or cytotoxic treatment seems most appropriate to adequately assess the role of these agents in secondary cancer growth prevention. These adjuvant studies will, by their design, definitely be time and patient consuming, and here an important as yet unanswered question is that of patient convenience and optimal duration of treatment.
In conclusion, 5 years of clinical studies with antiangiogenic agents have yielded promising results, and therefore antiangiogenic agents have the potential to play a role as active anticancer agents. To definitely confirm this role, specifically designed phase I studies, using the correct end points, must be able to define one or more safe and biologically active doses that can be tested in properly designed randomised phase II trials. Here, a wide array of possible combinations can be thought of. Finally, it is mandatory that in more than one randomised phase III trial patient benefit is demonstrated.
The role of the VTAs is not yet established as only phase I studies have been performed so far, but considering their mechanism of action, more studies will definitely follow.
After a period of flawing interest, antiangiogenic agents have regained their place in the centre of investigational antitumour treatment, and it is without any doubt that in the future their role in clinical practice will be established.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Beerepoot L, Witteveen P, Groenewegen G, Fogler W, Sim B, Sidor C, Phillips E, Zonnenberg B, Schramel F, Gebbink M, Voest E (2001) Preliminary results of a phase I trial of recombinant human angiostatin by twice-daily subcutaneous injection in patients with advanced cancer. Proc AACR-NCI-EORTC (abstract 34)
Bhargava P, Marshall J, Rizvi N, Dahut W, Yoe J, Figuera M, Phipps K, Ong V, Kato A, Hawkins M (1999) A phase I and pharmacokinetic study of TNP-470 administered weekly to patients with advanced cancer. Clin Cancer Res 5: 1989–1995
Blumenschein G, Fossella F, Pisters K, Khuri F, Lu C, Kies M, Glisson B, Zinner R, Crane E, Schaerer R, Dordal M, Goodin T, Hong W, Herbst R (2002) A phase I study of TNP-470 continuous infusion alone or in combination with paclitaxel and carboplatin in adult patients with NSCLC and other solid tumors. Proc Am Soc Clin Oncol 21: 314a (abstract 1254)
Brahmer J, Kelsey S, Scigalla P, Hill G, Bello C, Elza-Brown K, Donehower R (2002) A phase I study of SU6668 in patients with refractory solid tumors. Proc Am Soc Clin Oncol 21: 84a (abstract 335)
Britten C, Rosen L, Kabbinavar F, Rosen P, Mulay M, Hernandez L, Brown J, Bello C, Kelsey S, Scigalla P (2002) Phase I trial of SU6668, a small molecule receptor tyrosine kinase inhibitor, given twice daily in patients with advanced cancers. Proc Am Soc Clin Oncol 21: 28b (abstract 1922)
Burnstein H, Parker L, Savoie J, Younger J, Kuter I, Ryan P, Garber J, Campos S, Shulman L, Harris L, Gelman R, Winer E (2002) Phase II trial of the anti-VEGF antibody bevacizumab in combination with vinorelbine for refractory advanced breast cancer. Breast Cancer Res Treat 76(Suppl 1): S115 76: 115a (abstract 446)
De Vos F, Hoekstra R, Gietema J, Eskens F, van der Gaast A, Carr R, Glad-Anderson S, Humerickhouse R, Groen H, de Vries E, Verweij J (2002) A phase I dose escalating study of the angiogenesis inhibitor thrombospondinmimetic (ABT-510) in patients with advanced cancer. Proc Am Soc Clin Oncol 21: 82a (abstract 324)
DelProposto Z, LoRusso P, Latif Z, Morton P, Wheeler C Barge A, Evelhoch J (2002) MRI evaluation of the effects of vascular targeting agent ZD6126 on tumor vasculature. Proc Am Soc Clin Oncol 21: 111a (abstract 440)
DeMoraes E, Fogler W, Grant D, Wahl M, Leeper D, Zrada S, Malin A, Connors S, Fortier A, Dabrow M, Sidor C, Capizzi R (2001) Recombinant human angiostatin (rhA): a phase I clinical trial assessing safety, pharmacokinetics (PK) and pharmacodynamics (PD). Proc Am Soc Clin Oncol 20: 3a (abstract 10)
Dowlati A, Haaga J, Remick SC, Spiro TP, Gerson SL, Liu L, Berger SJ, Berger NA, Willson JK (2001) Sequential tumor biopsies in early phase clinical trials of anticancer agents for pharmacodynamic evaluation. Clin Cancer Res 7: 2971–2976
Dowlati A, Robertson K, Cooney M, Petros WP, Stratford M, Jesberger J, Rafie N, Overmoyer B, Makkar V, Stambler B, Taylor A, Waas J, Lewin JS, McCrae KR, Remick SC (2002) A phase I pharmacokinetic and translational study of the novel vascular targeting agent combretastatin a-4 phosphate on a single-dose intravenous schedule in patients with advanced cancer. Cancer Res 15: 3408–3416
Drevs J, Mross K, Medinger M, Mueller M, Laurent D, Reitsma D, Henry A, Xia J, Marme D, Unger C (2003) Phase I dose escalation and pharmacokinetic study of the VEGF inhibitor PTK787/ZK222584 in patients with liver metastases. Proc Amer Soc Clin Oncol 22: 284 (abstract 1142)
Drevs J, Schmidt-Gersbach C, Mross K, Medinger M, Mueller M, Steward W, Laurent D, Lee L, Dugan M, Henry A, Marme D, Unger C (2002) Surrogate markers for the assessment of biological activity of the VEGF-receptor inhibitor PTK787/ZK222584 (PTK/ZK) in two clinical phase I trials. Proc Am Soc Clin Oncol 21: 85a (abstract 337)
Dupont J, Camastra D, Gordon M, Mendelson D, Murren J, Hsu A, Lucarelli A, Cedarbaum J, Spriggs D (2003) A phase I study of VEGF trap in patients with solid tumors and lymphoma. Proc Am Soc Clin Oncol 22: 194 (abstract 776)
Eder J, Supko J, Clark J, Puchalski T, Garcia-Carbonero R, Ryan D, Shulman L, Proper J, Kirvan M, Rattner B, Connors S, Keogan M, Janicek M, Fogler W, Schnipper L, Kinchla N, Sidor C, Phillips N, Folkman J, Kufe D (2002) Phase I clinical trial of recombinant human endostatin administered as a short intravenous infusion repeated daily. J Clin Oncol 20: 3772–3784
Eskens F, Dumez H, Hoekstra R, Perschl A, Brindley C, Bottcher S, Wynendaele W, Drevs J, Verweij J, van Oosterom A (2003) Phase I and pharmacokinetic study of continuous twice weekly intravenous administration of cilengitide (EMD 121974), a novel inhibitor of the integrins alphavbeta3 and alphavbeta5 in patients with advanced solid tumours. Eur J Cancer 39: 917–926
Faivre S, Chieze S, Marty M, Hammershaimb L, Pluda J, Lozahic S, Armand J, Raymond E (2003) Safety profile and pharmacokinetic analysis of medi-522, a novel humanized monoclonal antibody that targets integrin αvβ3 receptor, in patients with refractory solid tumors. Proc Am Soc Clin Oncol 22: 208 (abstract 832)
Gadgeel S, LoRusso P, Wozniak A, Wheeler C (2002) A dose-escalation study of the novel vascular targeting agent ZD6126 in patients with solid tumors. Proc Am Soc Clin Oncol 21: 110a (abstract 438)
Galbraith S, Rustin G, Lodge M, Taylor N, Stirling J, Jameson M, Thompson P, Hough D, Gumbrell L, Padhani A (2002) Effects of 5,6-dimethylxanthenone-4-acetic acid (DMXAA) on human tumour microcirculation assessed by dynamic enhanced magnetic resonance imaging (DCE-MRI). J Clin Oncol 20: 3826–3840
Gordon MS, Margolin K, Talpaz M, Sledge Jr GW, Holmgren E, Benjamin R, Stalter S, Shak S, Adelman D (2001) Phase I safety and pharmacokinetic study of recombinant human anti-vascular endothelial growth factor in patients with advanced cancer. J Clin Oncol 19: 843–850
Gordon MS, Mendelson D, Guirguis M, Knight R, Humerickhouse R, Stopeck A, Wang Q (2003) ABT-510, an anti-angiogenic, thrombospondin-1 (TSP-1) mimetic peptide, exhibits favorable safety profile and early signals of activity in a randomized phase 1b trial. Proc Am Soc Clin Oncol 22: 195 (abstract 780)
Gutheil J, Campbell T, Pierce P, Watkins J, Huse W, Bodkin D, Cheresh D (2000) Targeted antiangiogenic therapy for cancer using Vitaxin: a humanized monoclonal antibody to the integrin alphavbeta3. Clin Cancer Res 6: 3056–3061
Hansma A, Hoekman K, Broxterman H, Kievit E, van der Horst I, Boven E, Schalkwijk C, Pinedo H (2002) A phase I study of rhEndostatin: continuous intravenous (iv) followed by subcutaneous (sc) administration. Proc Am Soc Clin Oncol 21: 110a (abstract 436)
Herbst R, Hess K, Tran H, Tseng J, Mullani N, Charnsangavej C, Madden T, Davis D, McConkey D, O'Reilly M, Ellis L, Pluda J, Hong W, Abbruzzese J (2002) Phase I study of recombinant human endostatin in patients with advanced solid tumors. J Clin Oncol 20: 3792–3803
Holden S, Morrow M, O'Bryant C, Pluda J, Eckhardt S (2002) Correlative biological assays to guide dose escalation in a phase I study of the antiangiogenic αVβ3 and αVβ5 integrin antagonist EMD121974 (EMD). Proc Am Soc Clin Oncol 21: 28a (abstract 110)
Hurwitz H, Fehrenbacher L, Cartwright T, Hainsworth J, Heim W, Berlin J, Griffing S, Novotny W, Holmgren E, Kabbinanvar F (2003) Bevacizumab (a monoclonal antibody to vascular endothelial growth factor) prolongs survival in first-line colorectal cancer (CRC): results of a phase III trial of bevacizumab in combination with bolus IFL (irinotecan, 5-fluorouracil, leucovorin) as first-line therapy in subjects with metastatic CRC. Proc Am Soc Clin Oncol (abstract 3646)
Hurwitz H, Holden S, Eckhardt S, Rosenthal M, de Boer R, Rischin D, Green M, Basser R (2002) Clinical evaluation of ZD6474, an orally active inhibitor of VEGF signaling, in patients with solid tumors. Proc Am Soc Clin Oncol 21: 82a (abstract 325)
Kabbinavar F, Hurwitz HI, Fehrenbacher L, Meropol NJ, Novotny WF, Lieberman G, Griffing S, Bergsland E (2003) Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol 21: 60–65
Klagsbrun M, D' Amore P (1996) Vascular endothelial growth factor and its receptors. Cytokine Growth Factor Rev 7: 259–270
Kuenen B, Rosen L, Smit E, Parson M, Levi M, Ruijter R, Huisman H, Kedde M, Noordhuis P, van der Vijgh W, Peters G, Cropp G, Scigalla P, Hoekman K, Pinedo H, Giaccone G (2002a) Dose-finding and pharmacokinetic study of cisplatin, gemcitabine, and SU5416 in patients with solid tumors. J Clin Oncol 20: 1657–1667
Kuenen B, Ruijter R, Hoekman K, Scigalla P, Giaccone G, Pinedo H (2002b) Dose finding study of SU6668 given thrice daily by oral route under fed conditions in patients with advanced malignancies. Proc Am Soc Clin Oncol 21: 110a (abstract 437)
Langmuir V, Cobleigh M, Herbst R, Holmgren E, Hurwitz H, Kabbinavar F, Miller K, Novotny W (2002) Successful long-term therapy with bevacizumab (Avastin) in solid tumors. Proc Am Soc Clin Oncol 21: 9a (abstract 32)
Logothetis C, Wu K, Finn L, Daliani D, Figg W, Ghaddar H, Gutterman J (2001) Phase I trial of the angiogenesis inhibitor TNP-470 for progressive androgen-independent prostate cancer. Clin Cancer Res 7: 1198–1203
Manning W, Bello C, Deprimo S, Fletcher J, Fletcher C, Van Den Abbeele A, Cohen D, Scigalla P, Cherrington J, Demetri G (2003) Pharmacokinetic and pharmacodynamic evaluation of SU11248 in a phase I clinical trial of patients (pts) with imitanib-resistant gastrointestinal stromal tumor (GIST). Proc Am Soc Clin Oncol 22: 192 (abstract 768)
Margolin K, Gordon MS, Holmgren E, Gaudreault J, Novotny W, Fyfe G, Adelman D, Stalter S, Breed J (2001) Phase Ib trial of intravenous recombinant humanized monoclonal antibody to vascular endothelial growth factor in combination with chemotherapy in patients with advanced cancer: pharmacologic and long-term safety data. J Clin Oncol 19: 851–856
Mendel D, Laird A, Xin X, Li G, Schreck R, Carver J, Louie S, Sukbuntherng J, Plise E, Kelsey S, Scigalla P, Cherrington J (2002) Development of a preclinical pharmacokinetic/pharmacodynamic relationship for the angiogenesis inhibitor SU11248, a selective inhibitor of VEGF and PDGF receptor tyrosine kinases in clinical development. Proc Am Soc Clin Oncol 21: (abstract 94)
Miller K (2003) E2100: a phase III trial of paclitaxel versus paclitaxel/bevacizumab for metastatic breast cancer. Clin Breast Cancer 3: 421–422
Miller K, Rugo H, Cobleigh M, Marcom P, Chap L, Holmes F, Fehrenbacher L, Overmoyer B, Reimann J, Vassel A, Langmuir V (2002) Phase III trial of capecitabine (Xeloda) plus bevacizumab (Avastin) versus capecitabine alone in women with metastatic breast cancer (MBC) previously treated with an anthracycline and a taxane. Breast Cancer Res Treat 76(Suppl 1): S37 (abstract 36)
Minami H, Ebi H, Tahara M, Sasaki Y, Yamamoto M, Yamamda Y, Tamura T (2003) A phase I study of an oral VEGF receptor tyrosine kinase inhibitor ZD6474 in Japanese patients with solid tumors. Proc Am Soc Clin Oncol 22: 194 (abstract 778)
Patel S, Jenkins J, Papadopolous N, Burgess M, Plager C, Gutterman J, Benjamin R (2001) Pilot study of vitaxin – an angiogenesis inhibitor – in patients with advanced leiomyosarcomas. Cancer 92: 1347–1348
Posey J, Ng T, Yang B, Khazaeli M, Carpenter M, Fox F, Needle M, Waksal H, LoBuglio A (2003) A phase I study of anti-kinase insert domain-containing receptor antibody, IMC-1C11, in patients with liver metastases from colorectal carcinoma. Clin Cancer Res 9: 1323–1332
Radema S, Beerepoot L, Witteveen P, Gebbink M, Wheeler C, Voest E (2002) Clinical evaluation of the novel vascular targeting agent ZD6126: assessment of toxicity and surrogate markers of vascular damage. Proc Am Soc Clin Oncol 21: 110a (abstract 439)
Raymond E, Faivre S, Vera K, Delbaldo C, Robert C, Spatz A, Bello C, Brega N, Scigalla P (2003) Final results of a phase I and pharmacokinetic study of SU11248, a novel multi-target tyrosine kinase inhibitor in patients with advanced cancers. Proc Am Soc Clin Oncol 22: 192 (abstract 769)
Rosen L, Mulay M, Long J, Wittner J, Brown J, Marino AM, Bello C, Walter S, Scigalla P, Zhu J (2003) Phase I trial of SU11248, a novel tyrosine kinase inhibitor in advanced solid tumors. Proc Am Soc Clin Oncol 22: 191 (abstract 765)
Rosen L, Rosen P, Kabbinavar F, Mulay M, Mickey J, Hernandez L, Brown J, Alexander J, Bello C, Cropp G, Kelsey S, Scigalla P (2001) Phase I experience with SU6668, a novel multiple receptor tyrosine kinase inhibitor in patients with advanced malignancies. Proc Am Soc Clin Oncol 20: 97a (abstract 383)
Rustin G, Bradley C, Galbraith S, Stratford M, Loadman P, Waller S, Bellenger K, Gumbrell L, Folkes L, Halbert G (2003) 5,6-dimethylxantenone-4-acetic acid (DMXAA), a novel antivascular agent: phase I clinical and pharmacokinetic study. Br J Cancer 88: 1160–1167
Rustin G, Price P, Stratford M, Galbraith S, Anderson H, Folkes L, Robbins A, Senna L (2001) Phase I study of weekly intravenous combretastatin A4 phosphate (CA4P): pharmacokinetics and toxicity. Proc Am Soc Clin Oncol 20: 99a (abstract 392)
Stadler W, Kuzel T, Shapiro C, Sosman J, Clark J, Vogelzang N (1999) Multi-institutional study of the angiogenesis inhibitor TNP-470 in metastatic renal carcinoma. J Clin Oncol 17: 2541–2545
Stevenson J, Gallagher M, Sun W, Algazy K, Vaughn D, Haller D, Hiller K, Halloran L, O'Dwyer P (2000) Phase I/pharmacokinetic trial of the endothelial toxin combretastatin A4P (CA4P) administered as an iv bolus on a daily X5 schedule every 21 days. Proc Am Assoc Cancer Res 11: 544 (abstract 3469)
Steward W, Thomas A, Morgan B, Wiedenmann B, Bartel C, Vanhoefer U, Trarbach T, Riedel U, Laurant D, Reitsma D (2003) Extended phase I/II study of the oral vascular endothelial growth factor (VEGF) receptor inhibitor PTK787/ZK222854 in combination with oxaliplatin/5-fluorouracil/leucovorin as first-line treatment for metastatic colorectal cancer. Proc Am Soc Clin Oncol 22: 274 (abstract 1098)
Stopeck A, Sheldon M, Vahedian M, Cropp G, Gosalia R, Hannah A (2003) Results of a phase I dose-escalating study of the antiangiogenic agent, SU5416, in patients with advanced malignancies. Clin Cancer Res 8: 2798–2805
Thomas A, Morgan B, Drevs J, Jivan A, Buchert M, Horsfield M, Hennig J, Mross K, Henry K, Ball H, Peng B, Fuxius S, Unger S, O'Byrne K, Laurent D, Dugan M, Steward W (2001) Pharmacodynamic results using dynamic contrast enhanced magnetic resonance imaging, of 2 phase 1 studies of the VEGF inhibitor PTK787/ZK 222584 in patients with liver metastases from colorectal cancer. Proc Am Soc Clin Oncol 20: 71a (abstract 79)
Tolcher A, Forero L, Celio P, Hammond L, Patnaik A, Hill M, Verat-Follet C, Haacke M, Besenval M, Rowinsky E (2003) Phase I, pharmacokinetic and DCE-MRI correlative study of AVE6082A, an antivascular combretastatin analogue, administered weekly for 3 weeks every 28 days. Proc Am Soc Clin Oncol 22: 208 (abstract 834)
Tolcher A, Karp D, O'Leary J, DeBono J, Caulkins J, Molpus K, Sutula K, Ferrante K, Gualberto A, Noe D, Huberman M, Rowinsky E (2002) A phase I and biological correlative study of an oral vascular endothelial growth factor receptor-2 (VEGFR-2) tyrosine kinase inhibitor, CP-547,632, in patients with advanced solid tumors. Proc Am Soc Clin Oncol 21: 84a (abstract 334)
Toner G, Mitchell P, De Boer R, Gibbs P, Hicks R, Scott A, McArtur G, Brega N, Massimini G, Scigalla P (2003) PET imaging study of SU112248 in patients with advanced malignancies. Proc Am Soc Clin Oncol 22: 191 (abstract 767)
Tran H, Herbst R, Xiong H, Madden T, Mulani N, Faria S, Kim K, Charnsangavej C, O'Reilly M, Abbruzzese J (2002) Phase I trial of recombinant human endostatin (rHu-endo) administered by continuous infusion (CI) intravenously (IV) in patients with solid tumors: a preliminary report. Eur J Cancer 38(Suppl 7): S73 22: 285 (abstract 230)
Trarbach T, Schleucher N, Riedel U, Tewes M, Souppart C, Reitsma D, Seeber S, Laurant D, Vanhoefer U (2003) Phase I/II study of the oral vascular endothelial growth factor (VEGF) receptor inhibitor PTK787/ZK222854 in combination with irinotecan/5-fluorouracil/leucovorin in patients with metastatic colorectal cancer. Proc Am Soc Clin Oncol 22: 285 (abstract 1144)
Ueda Y, Shimoyama T, Murakami H, Yamamoto N, Yamada Y, Tamura T (2002) Phase I study of TSU-68, VEGF receptor tyrosine kinase inhibitor, by twice daily oral administration between meals in patients with advanced solid tumors. Proc Am Soc Clin Oncol (abstract 443) 21: 111a
Voest E, Beerepoot L, Groenewegen G, Fogler W, Kim Lee Sim B, Sidor C, Phillips E, Zonnenberg B, Schramel F, Gebbink M, Witteveen P (2002) Phase I trial of recombinant human angiostatin by twice-daily subcutaneous injection in patients with advanced cancer. Proc Am Soc Clin Oncol 21: 81a (abstract 322)
Yang J, Haworth L, Steinberg S, Rosenberg S, Novotny W (2002) A randomized double-blind placebo-controlled trial of bevacizumab (anti-VEGF antibody) demonstrating a prolongation in time to progression in patients with metastatic renal cancer. Proc Am Soc Clin Oncol 21: 54 (abstract 15)
Yung W, Friedman H, Conrad C, Reardon D, Provencale J, Jackson E, Leeds N, Serrajudin H, Laurant D, Reitsma D (2003) A phase I trial of single-agent PTK787/ZD222854 (PTK/ZK), an oral VEGFR tyrosine kinase inhibitor, in patients with recurrent glioblastoma multiforme. Proc Am Soc Clin Oncol 22: 99 (abstract 395)
Yung W, Friedman H, Jackson E, Provenzale J, Leeds N, Conrad C, Walker A, Henry A, Huang J, Laurent D, Dugan M (2002) A phase I trial of PTK787/ZD22854, a novel oral VEGFR TK inhibitor in recurrent glioblastoma. Proc Am Soc Clin Oncol 21: 79a (abstract 315)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Eskens, F. Angiogenesis inhibitors in clinical development; where are we now and where are we going?. Br J Cancer 90, 1–7 (2004). https://doi.org/10.1038/sj.bjc.6601401
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6601401
Keywords
This article is cited by
-
Newly synthesized methionine aminopeptidase 2 inhibitor hinders tumor growth
Drug Delivery and Translational Research (2023)
-
Pretreatment PET/CT imaging of angiogenesis based on 18F-RGD tracer uptake may predict antiangiogenic response
European Journal of Nuclear Medicine and Molecular Imaging (2019)
-
Overexpression of Trps1 contributes to tumor angiogenesis and poor prognosis of human osteosarcoma
Diagnostic Pathology (2015)
-
Scl gene construction, expression and effect on hemangioma
Molecular Biology Reports (2013)
-
Regulation of Vascular Endothelial Growth Factor Signaling by miR-200b
Molecules and Cells (2011)
