
Abstract
We report on a new duplication case of 7q11.23, reciprocal of the Williams–Beuren (WB) deletion. The patient, a 13-year-old girl, was ascertained within an array-CGH screening of patients with epilepsy and neuronal migration defects. Similarly to the first reported patient, she showed serious difficulties in expressive language in the absence of severe mental retardation and marked dysmorphic features. Magnetic resonance imaging (MRI) of the brain revealed an abnormal development of the cerebral cortex in the left temporal lobe, which showed a simplified gyral pattern, and increased cortical thickness. This finding, which might explain poor language development, suggests that the WB critical region might harbour a dosage-sensitive gene controlling the molecular machinery of neuronal migration, with regional specificity and lateralization. It will be important to confirm our findings in newly diagnosed patients with dup(7)(q11.23). We expect to detect many more patients with the same duplication using widespread clinical implementation of high-resolution genome analysis.
Similar content being viewed by others


Introduction
The biological bases of Williams syndrome are instructive for several reasons: firstly, the 7q11.23 cryptic deletion is mediated by homologous segmental duplications that are well known to confer genomic instability (see Antonell et al1 for a review of the evolutionary mechanisms shaping the genomic structure at 7q11.23). Secondly, in at least one-third of the cases, the deletion is the recombinant product of a cryptic paracentric inversion in the parent of origin.2, 3 Finally, the phenotype results from a classical contiguous gene syndrome with elastin (ELN) haploinsufficiency responsible for the supravalvular aortic stenosis and haploinsufficiency of genes such as LIMK1, CYLN2 and GTF2IRD1 possibly linked to the cognitive and craniofacial pathology.4 For years it had been hypothesized that the 7q11.23 duplication, which is the reciprocal product of the same non-allelic homologous recombination associated with the deletion, had to be present either in persons with a specific abnormal phenotype or even in normal persons. The lack of such a finding led us to speculate that the duplication was likely to be either associated with a very mild phenotype or non-syndromic. After a tantalizing wait, in the last few months three cases with dup(7)(q11.23) have been reported, one of which was in a patient with mild mental retardation and severe expressive-language delay.5 The remaining two were in a family including a boy with craniosynostosis and moderate language disability and his healthy father.6 Here, we report a fourth case, detected through an array-CGH screening of patients with epilepsy and neuronal migration defects. The parents of the proband gave consent for publication, inclusive of the patient's photos.
Clinical report
Physical examination and neuroimaging
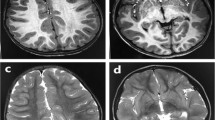
The proband (1574/2005) is the first child of healthy unrelated parents. The second pregnancy ended in the 12th week with a spontaneous abortion that resulted in a mosaic karyotype 45,XY,der(14;21)(q10;q10)/46,XY,+21,der(14;21)(q10;q10). The third pregnancy led to the birth of a normal 46,XX child. The proposita was born in the 40th week after spontaneous delivery weighing 3350 g (75th percentile), measuring 46 cm (10th percentile) in length and with a cranial circumference (OFC) of 34 cm (50th percentile). The Apgar was 9 and 10 at the first and fifth minute. No history of pre- or perinatal injury was mentioned. She was breast-fed for 6 months and had a regular sleep–wake rhythm. At birth, talipes equino-varus and hip dysplasia were corrected by prosthetic shoes and divaricators, respectively. Neuromotor development was reported to be normal. Pictures at 6 months of age (Figure 1a) revealed low-set ears with mild posterior rotation. At 4 years of age (Figure 1b), owing to severe speech and language delay (see below), audiometric, electroencephalographic (EEG) and karyotype investigations were performed, all yielding normal results. At the age of 12 years she presented her first partial seizure, which is why she was brought to our attention. Clinical examination revealed a girl with a round face and a stocky, short neck (Figure 1c–e), a dorsal hunch with kyphosis and abdominal obesity. She was 153 cm (25th–50th centile) tall, she weighed 53 kg (75th–90th centile), and her OFC was 56 cm (>2 SD). She wore corrective glasses for astigmatism (left eye with exotropia). We noted low-set, slightly posteriorly rotated ears, short philtrum and thin lips that, when she smiled, revealed an asymmetric opening of the mouth (Figure 1e). Her tongue showed a horizontal crease without invagination. Café-au-lait spots were present over the right nipple and at the sacral-coccygeal region. We also noted diffuse hirsutism (she was at Tanner stage 3–4). She had right cubitus valgus, stocky fingers and bilateral club foot. The electrocardiogram showed normal findings, as did abdominal, renal and cardiac ultrasound investigations. The magnetic resonance imaging (MRI) of the brain, performed at 13 years of age, revealed an abnormal development of the cerebral cortex in the left temporal lobe, which showed a simplified gyral pattern with poorly developed superior and middle temporal gyri on the lateral aspect and increased cortical thickness in the mesiobasal portion and in the insular cortex (Figure 2). The karyotype analysis performed twice in two different laboratories gave normal results.

The proband at different ages. (a) At 6 months. (b) At 4 years. (c) At 12 years. (d, e) At 13 years, a slightly asymmetric opening of the mouth is visible in (e).

MRI of the brain. T1-weighted images. (a, b) Coronal sections; (c) an axial section. (a) The left temporal lobe, represented here in its anterior part, is smaller with respect to the contralateral and shows a simplified gyral pattern. The superior and inferior temporal gyri are shallow (white arrows on the right) and the hippocampal gyrus is thickened (white arrow on the left). The overlying insular cortex is also thickened. (b) This section is contiguous to the previous (a). The two black arrows indicate areas of thickening in the hippocampal gyrus and in the inferior temporal gyrus. (c) On the axial plan, it is apparent how the areas of cortical thickening in the mesial and lateral temporal lobe, correspond to regions of abnormal cortical infolding (white arrows).
Neurological and psychiatric evaluation
On neurologic examination, when she was 12 years old, she was noted to be left-handed and to have mild dysmetria with tandem gait and unipedal stance. At this age, partial seizures were characterized by unresponsiveness, lip cyanosis, clonic jerks on the right hemibody with head deviation to the right. These seizures lasted 1–3 min and were followed by sleep. Initially, about two seizures a month occurred in spite of treatment with topiramate and valproic acid, but after a few months seizure control was obtained. Several EEGs showed sharp waves on the left temporal region. Her speech and language had always been very poor. At 8 years old, she was only able to pronounce bisillabic words with phonological errors (d/t, r/l and r/gl substitution) without deficit of orofacial praxis as demonstrated by testing. At the age of 12 years, she could pronounce bisillabic sounds to answer questions and a few very simple sentences limited to her daily activities. Memory, learning and attention assessed with the test of memory and learning revealed low scores (<16th centile). Lexical comprehension abilities were examined through the peabody picture vocabulary test (PPVT)-III, revealing a decrease in her functioning (IQ=65 at PPVT). Deficit in discrimination were further documented with the token test (<5 SD). The general cognitive ability assessed with the WISC-R revealed a full-scale IQ of 53 (verbal: 51; performance: 58). Visual motor integration test revealed poor visuo-spatial skills with a score of 75 (<5 centile). Memory evaluation showed discrepancy between verbal (<1st centile) and visuo-spatial (<9th centile) span. The patient exhibited neither attention deficit or hyperactivity traits, nor sleep disorders, nor autistic-like behaviour.
On the whole, the girl was judged to have moderate mental retardation with severely impaired speech, and poor scoring in all the entry/exit battery of tests. Her social interaction capabilities were good: she appeared as a naïve, loving, quiet girl who liked to spend time playing with and taking care of her 2-year-old sister.
Materials and methods
Molecular karyotyping was performed through array-CGH with the Agilent kit. This platform is a 60-mer oligonucleotide-based microarray that allows genome-wide survey and molecular profiling of genomic aberrations with a resolution of ∼75 kb (kit 44B). DNAs (7 μg) of the patient and controls of the same sex (Promega) were double-digested with RSAI and AluI for 2 h at 37°. After columns purification, 2 μg of each digested sample were labelled by random priming (Invitrogen) for 2 h using Cy5-dUTP for the patients/parents DNA and Cy3-dUTP for the control DNA. Labelled products were columns purified. After probes denaturation and pre-annealing with 50 μg of Cot-1 DNA, hybridization was performed at 65° with rotation for 40 h. After two washing steps, the array was analysed through the Agilent scanner and the Feature Extraction software (v8.1). Graphical overview was obtained using the CGH analytics software (v3.2.32).
Genotyping of polymorphic loci on the proband and the parental DNAs was performed by amplification with primers labelled with fluorescent probes (ABI 5-Fam and Hex) followed by analysis on an ABI 310 Genetic Analyzer (Applied Biosystems).
FISH was performed on metaphase and interphase cells from the proposita and her mother to investigate if the duplication was direct or inverted and if it was mediated by a paracentric inversion. To this end, BAC clones allocated in the duplication region (RP11-815K13 proximal to the critical region, CTA-208H19 and RP5-1186P10 both within the critical region2) were used. DNAs extraction, labelling, hybridization and detection and FISH experiments were undertaken as described previously.7
Analysis of the breakpoints with self-chain and segmental duplications repeats tools from UCSC Genome Bioinformatics (http://genome.ucsc.edu/) has also been performed.
Results
Array-CGH demonstrated a duplication of at least 1.438 Mb ranging from 72.145 to 73.583 Mb flanked by oligomeres at 71.846 and 73.926 that were not duplicated (Figure 3a). Microsatellite analysis demonstrated the maternal origin of the duplication with three alleles, two maternal and one paternal at D7S3194 and D7S1870 (Figure 3b). FISH analysis demonstrated that the duplication was in tandem (Figure 3c) and that the mother was not inverted at the duplication region (Figure 3d).

(a) Array-CGH profile of chromosome 7 showing the duplication at 7q11.23. On the right, the enlargement of the duplicated region: probes from 72.145 to 73.583 Mb have an average log2 ratio of about 0.56, compatible with duplication; (b) microsatellite analysis with STS D7S3194 showing three alleles in the proposita, two of them of maternal origin; (c) interphase FISH in the proposita showing that clones CTA-208H19 and RP5-1186P10 (in red and green, respectively) are duplicated in tandem (black arrow); RP11-815K13, in yellow, is in single copy because is outside the critical region; white arrow indicates the normal chromosome 7; (d) interphase FISH in the proposita's mother showing that RP11-815K13 (yellow), CTA-208H19 and RP5-1186P10 both within the critical region (red and green, respectively) have the same order in both chromosomes (white arrows).
Discussion
The rearrangement
The WBS deletion is mediated by non-allelic homologous recombination between homologous blocks of segmental duplications that have emerged over the past ∼35 million years.1 Bayes et al3 demonstrated that in 90% of the cases, the non-allelic homologous recombination leading the deletion occurs between the centromeric and medial so-called B blocks. In our case, array-CGH demonstrated that the duplication breakpoints are the same as the common deletion with the proximal breakpoint (between 71.846 and 72.145 Mb) falling within the centromeric B block and the distal breakpoint (between 73.583 and 73.926 Mb) within the medial B block.3 Interphase FISH showed that the duplication was in tandem and that the mother, who transmitted the duplicated chromosome (Figure 3), was not inverted at the critical region.
The dup(7)(q11.23) phenotype
We report the phenotypic description of a girl with the 7q duplication reciprocal of the deletion associated with the Williams syndrome, who presented mild dysmorphic features, severe speech impairment and partial seizures. Two patients with the same duplication, as well as one of their fathers, had already been reported (Table 1).
Our patient has been studied in detail and, contrary to those already reported, showed no attention deficit or sleep disorders.5 Moreover, she did not have trigonocephalic synostosis as reported in the proband of Kriek et al.6 In our patient, a brain MRI showed abnormal cortical development in the left temporal lobe. Our patient, like the two described previously, indicates that the main clinical feature associated with dup(7)(q11.23) is speech and language delay, varying from moderate6 to severe, in the context of moderate mental retardation. As no hearing or anatomic defect of the phonoarticultory district were present in these patients, we may assume that the critical region contains a gene, the duplication of which results in brain abnormalities, in turn impairing language skills. The left temporal dysplasia revealed by MRI in our patient might indeed underlie poor language development. As the three previously reported patients did not undergo a brain MRI, it is not possible to know whether abnormal cortical development of the left temporal lobe was also present in them. If the father of the patient reported by Kriek et al6 had displayed normal speech, we might assume, as already noted,4 that, in addition to gene dosage, other mechanisms such as genetic and/or environmental interactions may be significant in determining the phenotype. It has been hypothesized that the 7q11.23 duplication phenotype is reciprocal to that of Williams syndrome, at least in the field of language development.4, 5 This observation would imply that the same gene is responsible for the ‘cocktail party’ pattern of speech in haploinsufficency and for the severe expressive-language absence if duplicated. If indeed the speech disorder of the 7q duplication is due to abnormal cortical development in cortical areas subserving language function, a gene similar to those known to control the molecular machinery of neuronal migration8 would be the most likely candidate. Two such genes, linked to microtubule-associated proteins, are present in the duplicated region: LIMK1, encoding a protein kinase which regulates actin filament dynamics, and CYLN2, encoding a protein presumably linking microtubules to dendritic lamellar body (UCSC Genome Bioinformatics). Recently, cortical dysplasia manifested as thickening of the right perisylvian cortex was documented in classical Williams syndrome,9 indeed reinforcing the idea that a dosage-sensitive gene, responsible for correct neuronal migration during embryogenesis, is present at 7q11.23. Genetically determined unilateral cortical malformations are relatively uncommon. Unilateral polymicrogyria was reported in patients with the diGeorge/velocardiofacial syndrome and deletion at chromosome 22q1110 and in two cousins with 1q44qter monosomy and 12p13.3pter trisomy secondary to a familial translocation.11 Recently, four families in which more than one individual had unilateral polymicrogyria were identified.12 It was hypothesized that candidate genes might have differential expression or function between the two hemispheres during cortical development.12 Whole mount in situ hybridization studies of LIMK1 and CYLN2 in mouse embryos would be necessary in order to confirm our hypothesis.
Language impairment in the absence of severe mental retardation and marked dysmorphic features has the main impact when identifying the dup(7)(q11.23) syndrome.
Finally, it is interesting to note that not only language absence is in contrast with the fluent language typical of the WBS deletion. However, macrocephaly (>2 SD) and normal stature in our patient are also in contrast with microcephaly and growth retardation in patients with Williams–Beuren syndrome.
References
Antonell A, de Luis O, Domingo-Roura X, Perez-Jurado LA : Evolutionary mechanisms shaping the genomic structure syndrome chromosomal region at human 7q11.23. Genome Res 2005; 15: 1179–1188.
Osborne LR, Li M, Pober B et al: A 1.5 million-base pair inversion polymorphism in families with Williams–Beuren syndrome. Nat Genet 2001; 29: 321–325.
Bayes M, Magano LF, Rivera N, Perez-Jurado LA : Mutational mechanisms of Williams–Beuren syndrome deletions. Am J Hum Genet 2003; 73: 131–151.
Tassabehji M, Donnai D : More or less? Segmental duplications and deletions in the Williams–Beuren syndrome region provide new insights into language development. Eur J Hum Genet 2006; 14: 507–508.
Somerville MJ, Mervis CB, Young EJ et al: Severe expressive-language delay related to duplication of the Williams–Beuren locus. N Engl J Med 2005; 353: 1694–1701.
Kriek M, White SJ, Szuhai K et al: Copy number variation in regions flanked (or unflanked) by duplicons among patients with developmental delay and/or congenital malformations, detection of reciprocal and partial Williams–Beuren duplications. Eur J Hum Genet 2006; 14: 180–189.
Ciccone R, Giorda R, Gregato G et al: Reciprocal translocations: a trap for cytogenetists? Hum Genet 2005; 117: 571–582.
Gressens P : Pathogenesis of migration disorders. Curr Opin Neurol 2006; 19: 135–140.
Thompson PM, Lee AD, Dutton RA et al: Abnormal cortical complexity and thickness profiles mapped in Williams syndrome. J Neurosci 2005; 25: 4146–4158.
Sztriha L, Gururaj A : Hippocampal dysgenesis associated with temporal lobe hypoplasia and arachnoid cyst of the middle cranial fossa. J Child Neurol 2005; 20: 926–930.
Zollino M, Colosimo C, Zuffardi O et al: Cryptic t(1;12)(q44;p13.3) translocation in a previously described syndrome with polymicrogyria, segregating as an apparently X-linked trait. Am J Med Genet A 2003; 117: 65–71.
Chang BS, Apse KA, Caraballo R et al: A familial syndrome of unilateral polymicrogyria affecting the right hemisphere. Neurology 2006; 66: 133–135.
Acknowledgements
This work was supported by the Telethon foundation (to TP: GGP05177) and PRIN 2005, Fondazione Mariani and Fondazione CARIPLO (all to O.Z.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Torniero, C., Bernardina, B., Novara, F. et al. Cortical dysplasia of the left temporal lobe might explain severe expressive-language delay in patients with duplication of the Williams–Beuren locus. Eur J Hum Genet 15, 62–67 (2007). https://doi.org/10.1038/sj.ejhg.5201730
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201730
Keywords
This article is cited by
-
High-resolution chromosomal microarray analysis for copy-number variations in high-functioning autism reveals large aberration typical for intellectual disability
Journal of Neural Transmission (2020)
-
Transcription Factor 2I Regulates Neuronal Development via TRPC3 in 7q11.23 Disorder Models
Molecular Neurobiology (2019)
-
The nuclear localization pattern and interaction partners of GTF2IRD1 demonstrate a role in chromatin regulation
Human Genetics (2015)
-
Brief Report: Functional MRI of a Patient with 7q11.23 Duplication Syndrome and Autism Spectrum Disorder
Journal of Autism and Developmental Disorders (2014)
-
Association of GTF2i in the Williams-Beuren Syndrome Critical Region with Autism Spectrum Disorders
Journal of Autism and Developmental Disorders (2012)
