
Abstract
Intellectual disability (ID) represents an extremely heterogeneous group of disorders, characterized by significant limitations in intellectual function and adaptive behavior. Among the monogenic causes, autosomal recessive genes (ARID) are responsible for more than 50% of ID. Here, we report a novel in-frame homozygous deletion variant [c.730_753del; p.(Ala244_Gly251del)] in SOX4 (sex-determining region Y-related high-mobility group box 4), segregating with moderate to severe ID, hypotonia, and developmental delay in a Pakistani family. Our identified variant p.(Ala244_Gly251del) is predicted to remove evolutionarily conserved residues from the interdomain region and may destabilize the protein secondary structure. SOX4 belongs to group C of the SOX transcription regulating family known to be involved in early embryo development. Single-cell RNA data analysis of developing telencephalon revealed highly overlapping expression of SOX4 with SOX11 and DCX, known neurogenesis regulators. Our study expands the mutational landscape of SOX4 and the repertoire of the known genetic causes of ARID.
Similar content being viewed by others


Introduction
Members of the SOX [Sex-determining region Y (SRY)-related high-mobility group (HMG) box] transcriptional regulation family play key roles in fetal development [1]. Approximately 20 members of the SOX family have been identified in mammalian species and are involved in organogenesis through appropriate progenitor cell production and differentiation [2]. SOX proteins have 50% sequence homology to SRY in the HMG domain that binds the minor grove of DNA [3, 4]. Thus, cell fate is controlled through transcriptional regulation of genes involved in embryonic development. Pathogenic variants in different SOX genes have been associated with various syndromes in humans. For instance, Coffin–Siris syndrome 9 (MIM: 615866), campomelic dysplasia (MIM: 608160), Lamb–Shaffer syndrome (MIM: 604975), and Waardenburg–Hirschsprung disease (MIM: 602229) are caused by pathogenic alleles of SOX11, SOX9, SOX5, and SOX10, respectively (Supplementary Table S1).
Previously, four de novo heterozygous missense variants in the HMG domain of SOX4 were associated with neurodevelopmental disorders with mild dysmorphism in four unrelated affected individuals [3]. Here, we report a novel homozygous in-frame deletion in SOX4 segregating with intellectual disability (ID), hypotonia, and developmental delay in a consanguineous Pakistani family PKMR225 (Fig. 1).

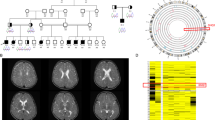
A Pedigree of family PKMR225. The filled symbols represent affected individuals, and a double horizontal line connecting parents represents a consanguineous marriage. Genotypes for the identified SOX4 variant are given for the participating individuals. B Photographs and T2-weighted MRI images of affected individuals (IV:1 and IV:2). Facial feature recognition analysis revealed mild facial dysmorphism in both individuals. Affected individual IV:1 has a long triangular face with deep-set eyes, micrognathia, and high palate, while affected individual IV:2 has an upslanted palpebral fissure, broad base nose, and diastema. Both affected individuals have normal hands and no brain tissue abnormality was noted in the MRI scans. C SOX4 has a single coding exon gene. Shown are all four reported de novo (black) variants and the novel variant c.730_753del (red) found in this study. D Evolutionary conservation of amino acids deleted due to the c.730_753del variant [Color code Red: Small (small + hydrophobic [includes aromatic –Y]), Blue: Acidic, Green: Hydroxyl + sulfhydryl + amine + G]. E Tolerance landscape visualization of SOX4 via MetaDome with relative positions of the four reported de novo missense and novel in-frame deletion variants. The graph indicates that variants within the HMG domain are “highly intolerant,” while the in-frame deletion outside the HMG domain is also “intolerant” for substitutions.
Subjects and methods
This study was approved by the Institutional Review Boards at the University of Maryland Baltimore, USA, and the Centre of Excellence in Molecular Biology, University of The Punjab, Lahore, Pakistan. Family PKMR225 was ascertained from Multan, Punjab province of Pakistan after obtaining the written informed consents. The affected individuals were examined by a neurologist and primary physician for specific features related to ID including neurological, morphological, behavioral, ophthalmological, auditory, skeletal, and dermatological abnormalities. Blood samples were collected for DNA extraction and biochemical assays.
Whole exome sequencing (WES) was performed on individual IV:2 DNA sample as reported previously [5]. Variants obtained from WES were filtered using the criteria previously described in [6]. The final SOX4 candidate variant was Sanger sequenced in all the participating family members. In silico analysis and three-dimensional (3D) molecular modeling were performed to assess the impact of the identified deletion on the encoded protein using the Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/), MetaDome (https://stuart.radboudumc.nl/metadome/), I-Tasser (https://zhanglab.ccmb.med.umich.edu/I-TASSER/), and PyMol programs. GTEx (https://www.gtexportal.org/) and UCSC cell browser (https://cells.ucsc.edu/) were used to analyze SOX4 and related genes expression levels in different brain regions and developing telencephalon, respectively.
Results
Detailed clinical presentations of the affected individuals are shown in Table 1. Briefly, the two affected individuals (IV:1 and IV:2) of family PKMR225 were born to consanguineous parents (Fig. 1A). Their mother had a history of two miscarriages and one induced abortion. Both affected individuals have had moderate and severe ID since childhood. Affected individual IV:1 has global developmental delay with aggressive behavior. He started sitting at the age of 2 years and independently walked at the age of 3 years. When given instruction, the individual was able to perform tasks, but showed socialization reluctance. Affected individual IV:2 started speaking at the age of 3 years and walking at the age of 4 years. She had diastema with a broad base nose and pale skin. She was not able to follow instructions. Magnetic resonance imaging (MRI) of both patients showed normal brain morphology (Fig. 1B). Both affected individuals had hypotonia and mild facial dysmorphism (Table 1). Serum electrolytes and complete blood count parameters for affected individual IV:2 were also within the normal range (Supplementary Table S2).
WES results from individual IV:2 unveiled SOX4 (accession no. NM_003107) as one of the candidate ID-causing gene. Sanger sequencing of all the available DNA samples confirmed segregation of a homozygous novel deletion variant (c.730_753delGCCGCCGCCCTGCTGCCCCTGGGC) of SOX4 among the two affected individuals, while the parents were both heterozygous (Fig. 1A). The identified variant is predicted to remove eight evolutionary conserved amino acids [p.(Ala244_Gly251del)] from the relatively intolerant region of SOX4 (Fig. 1D, E). Our 3D modeling of the mutant protein suggested that the loss of seven hydrogen bonds impacted the secondary structure and folding of SOX4 (Fig. 2A) and thus might impact the SOX4 DNA binding ability. In the wild-type protein, p.Ala234 is predicted to form hydrogen bonds with p.Ala246 (2.0 Å) and p.Leu247 (2.6 and 2.4 Å). Furthermore, p.Ser236 is also predicted to form two hydrogen bonds (2.2 and 2.1 Å) with p.Ala244, while p.Lys261 bonds (1.9 Å) with p.Ala246, and p.Ala252 bonds (1.9 Å) with p.Leu248. These hydrogen bonds would all be lost due to the p.(Ala244_Gly251del) variant found in family PKMR225, and thus, the deletion is predicted to negatively impact the protein conformation and ability to bind with other proteins or DNA.

A 3D SOX4 protein modeling. Hydrogen bonding between the residues is shown with dotted lines along with the distances in Å. At least seven predicted hydrogen bonds are lost due to the p. Ala244_Glu251 variant. B Single-cell RNA-seq visualization of SOX4 RNA expression with its interactors DCX, Sox11, TBR2, and WDR45 using UCSC cell browser for cortex development dataset, generated from expression of 4261 cells. Gene panels show expression data plotted in t-SNE on WGCNA layout, areas of interest are highlighted with orange lines, beige to dark brown show high RNA expression levels whereas blue shows absence of expression in developing human telencephalon. For cell type clustering details, see https://cells.ucsc.edu/?ds=cortex-dev. MGE medial ganglionic eminence, IPC intermediate progenitor cells.
Next, we analyzed the expression of SOX4 and known downstream targets in the publicly available databases of developing human brain tissues. High SOX4 expression was observed in telencephalon, particularly in interneurons, maturing excitatory neurons, and intermediate progenitor cells (IPCs) (Fig. 2B). Among the known targets, single-cell RNA (scRNA) data analysis revealed highly overlapping expression pattern of SOX4 with SOX11, and DCX in developing telencephalon (Fig. 2B). In mice, Sox4 has been reported to transactivate Tbr2 in the intermediate cortex progenitor cells [7]. Concordantly, we found highly overlapping expression of SOX4 and TBR2 in human IPCs (Fig. 2B). In contrast, we found low and diffuse expression of WDR45, also a known target of SOX4, in the developing telencephalon (Fig. 2B).
Discussion
Among the eight subgroups of the SOX family, three members, namely, SOX4, SOX11, and SOX12, constitute group C based on their high structural similarity in the HMG and transactivation domains [8]. SOX4 along with SOX11 is highly expressed in progenitor cells, and regulates several developmental processes in fetus. Besides proliferation, SOX4 also transactivates different genes, including DCX, TRB2, WDR45, for neurogenesis and other cellular processes [9,10,11]. DCX encodes a microtubule associated protein, which is involved in neuronal migration through tyrosine kinase signal transduction pathway [9]. T-box transcription factor (TRB2) induces neurogenesis indirectly through transit amplifying progenitors proliferation in the cortical subventricular zone [10], while, WDR45, also known to cause ID, participates in cellular autophagy pathway [11]. In contrast to other two members of group C, SOX12 has weak transcription activity. Loss of Sox12 in mice did not cause any apparent phenotype, due to functional compensation by Sox4 and Sox11 [12].
Our scRNA expression analysis in developing telencephalon supported involvement of SOX4 in neurogenesis through overlapping expression with SOX11 and DCX, known to be involved in neuronal cell division, maturation, and migration, respectively. Furthermore, TRB2 having high overlapping expression with SOX4 in IPCs have potency to develop into glutamatergic neurons. However, diffused expressional overlap of SOX4 and WDR45 can be due to spatiotemporal expression of WDR45 specific to autophagy. Inactivation studies suggest that Sox4 is involved in the maintenance of IPCs, whereas Sox11 is required for cell proliferation and differentiation, while coupled inactivation of both proteins showed impairment in neuronal cell survival and differentiation [13].
To date, four de novo heterozygous missense variants [p.(Ile59Ser), p.(Phe66Leu), p.(Lys105Asn), and p.(Ala112Pro)] in the highly intolerant region of the HMG domain (Fig. 2A, C) have been reported in index cases with Coffin–Siris 10 (MIM618506), which includes ID (Table 1). Among them, subjects with the p.(Ala112Pro) variant showed the most severe disease phenotype, including microcephaly, facial dysmorphism, weak muscle tone, tooth abnormalities, congenital vertical talus, and bilateral 5th finger clinodactyly, with no speech or walking ability (Table 1). Other features reported in these individuals include global developmental delay with facial, hand, and foot dysmorphism (Table 1). Similarly, the affected individuals of family PKMR225 had global developmental delay, moderate to severe ID, hypotonia, and mild facial dysmorphism (Table 1). However, these affected individuals neither have microcephaly nor clinodactyly, nor do they have a history of epilepsy. The parents, heterozygous for the p.(Ala244_Gly251del) variant, had normal IQ levels with no history of hypotonia, tooth abnormalities, or facial dysmorphism. The clinical findings suggest that in contrast to the previously reported de novo heterozygous variant, p.(Ala244_Gly251del) might be a hypomorphic allele of SOX4.
The previously reported de novo heterozygous missense variants are located in the highly conserved HMG domain of SOX4, while the in-frame deletion reported here is present in the uncharacterized interdomain region of SOX4 (Fig. 2D). Non-HMG domain sequences have the potential to affect the capability of encoded proteins to bind with other proteins or specific DNA regions. For instance, a previous study on SOX6 revealed essentiality of its non-HMG residues p.PLNLSSR for interaction with the corepressor protein CtBP2 [14]. Similarly, DNA-bound SOX9 requires residues on the carboxyl terminus to bind with HSP70 for the formation of transcriptional complexes [15]. These studies suggest that our identified deletion occurring in the non-HMG domain region has the potential to affect the transcriptional activity of the SOX4 protein by disrupting its protein or DNA binding sites.
Data availability
The variant has been submitted to ClinVar database (accession number: SCV001571323).
References
Laudet V, Stehelin D, Clevers H. Ancestry and diversity of the HMG box superfamily. Nucleic Acids Res. 1993;21:2493–501.
Koopman P, Schepers G, Brenner S, Venkatesh B. Origin and diversity of the SOX transcription factor gene family: genome-wide analysis in Fugu rubripes. Gene. 2004;328:177–86.
Zawerton A, Yao B, Yeager JP, Pippucci T, Haseeb A, Smith JD, et al. De novo SOX4 variants cause a neurodevelopmental disease associated with mild dysmorphism. Am J Hum Genet. 2019;104:246–59.
Wilson M, Koopman P. Matching SOX: partner proteins and co-factors of the SOX family of transcriptional regulators. Curr Opin Genet Dev. 2002;12:441–6.
Richard EM, Santos-Cortez RLP, Faridi R, Rehman AU, Lee K, Shahzad M, et al. Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss. Hum Mutat. 2019;40:53–72.
Riazuddin S, Hussain M, Razzaq A, Iqbal Z, Shahzad M, Polla D, et al. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol Psychiatry. 2017;22:1604–14.
Chen C, Lee GA, Pourmorady A, Sock E, Donoghue MJ. Orchestration of neuronal differentiation and progenitor pool expansion in the developing cortex by SoxC genes. J Neurosci. 2015;35:10629–42.
Hargrave M, Wright E, Kun J, Emery J, Cooper L, Koopman P. Expression of the Sox11 gene in mouse embryos suggests roles in neuronal maturation and epithelio‐mesenchymal induction. Developmental Dyn. 1997;210:79–86.
Ohshima T. Neuronal migration and protein kinases. Front Neurosci. 2015;8:458.
Lv X, Ren S-Q, Zhang X-J, Shen Z, Ghosh T, Xianyu A, et al. TBR2 coordinates neurogenesis expansion and precise microcircuit organization via Protocadherin 19 in the mammalian cortex. Nat Commun. 2019;10:1–15.
Cong Y, So V, Tijssen MA, Verbeek DS, Reggiori F, Mauthe M. WDR45, one gene associated with multiple neurodevelopmental disorders. Autophagy. 2021;12:1–16.
Hoser M, Potzner MR, Koch JM, Bösl MR, Wegner M, Sock E. Sox12 deletion in the mouse reveals nonreciprocal redundancy with the related Sox4 and Sox11 transcription factors. Mol Cell Biol. 2008;28:4675–87.
Dy P, Penzo-Mendez A, Wang H, Pedraza CE, Macklin WB, Lefebvre V. The three SoxC proteins—Sox4, Sox11 and Sox12—exhibit overlapping expression patterns and molecular properties. Nucleic Acids Res. 2008;36:3101–17.
Murakami A, Ishida S, Thurlow J, Revest J-M, Dickson C. SOX6 binds CtBP2 to repress transcription from the Fgf-3 promoter. Nucleic Acids Res. 2001;29:3347–55.
Marshall OJ, Harley VR. Identification of an interaction between SOX9 and HSP70. FEBS Lett. 2001;496:75–80.
Acknowledgements
We thank the PKMR225 family members for participating in this study. We would like to thank Muhammad Asad Usmani for his guidance and technical assistance.
Funding
This work was supported by National Institute of Neurological Disorders and Stroke, National Institutes of Health grant R01NS107428 to Saima Riazuddin.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was approved by the Institutional Review Boards (IRB) at the University of Maryland Baltimore, USA, and the Centre of Excellence in Molecular Biology (CEMB), University of The Punjab, Lahore, Pakistan.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ghaffar, A., Rasheed, F., Rashid, M. et al. Biallelic in-frame deletion of SOX4 is associated with developmental delay, hypotonia and intellectual disability. Eur J Hum Genet 30, 243–247 (2022). https://doi.org/10.1038/s41431-021-00968-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00968-w
This article is cited by
-
A new system for variant classification?
European Journal of Human Genetics (2022)
