
Abstract
Neurofibromatosis 1 predisposes affected individuals to the development of benign and malignant tumours that are frequently disfiguring and difficult to manage. However, advances in molecular biology and the development of mouse models have facilitated our understanding of disease pathogenesis. Positron emission tomography has demonstrated that sophisticated imaging techniques have a role in diagnosing complex problems like malignant peripheral nerve sheath tumours, while the prospect of targeted therapies for Nf1 complications is tantalisingly close.
Similar content being viewed by others

In brief
-
Neurofibromatosis 1 is an autosomal-dominant disorder with a prevalence of one in 4000–5000.
-
The major diagnostic features are café au lait patches, neurofibromas, skin-fold freckling, iris Lisch nodules, optic pathway glioma and bony dysplasia.
-
Neurofibromas are peripheral nerve sheath tumours comprising Schwann cells, fibroblasts, perineurial cells, mast cells, and axons in an extracellular matrix.
-
The Schwann cell initiates neurofibroma growth.
-
Cognitive impairment is the most common complication and presents with low average IQ, behavioural and specific learning problems.
-
There is 10% lifetime risk of developing malignant peripheral nerve sheath tumour (MPNST).
-
Symptoms of MPNST are persistent pain, rapid increase in size, change in texture and neurological deficit in association with a neurofibroma.
-
Vasculopathy including cardiovascular disease and cerebrovascular disease is a major cause of death in Nf1.
-
The gene for Nf1 is on chromosome 17q11.2; the gene product, neurofibromin acts as a tumour suppressor.
-
Neurofibromin has multiple functions including negative regulation of p21RAS, control of adenylyl cyclase activity and modulation of mTOR (mammalian target of rapamycin.)
Introduction
Neurofibromatosis 1, formerly termed von Recklinghausen's disease, is an autosomal dominant neurocutaneous disorder with a birth incidence of one in 2500 and a minimum prevalence of one in 4–5000.1 The Nf1 gene is located on chromosome 17q11.2 and the protein product termed neurofibromin acts as a tumour suppressor.2, 3, 4 The principal and defining manifestations of Nf1 are café au lait patches, neurofibromas (benign peripheral nerve sheath tumours), skin-fold freckling, iris Lisch nodules (hamartomas diagnosed on slit-lamp examination) and characteristic bony dysplasia of the long bones and sphenoid wing.5 The clinical expression and severity in Nf1 is diverse, even within families. The complications affect many of the body systems and range from disfigurement, scoliosis and vasculopathy to cognitive impairment and malignancy including peripheral nerve sheath tumours, and central nervous system gliomas. Macrocephaly, short stature and cutaneous angiomas are minor features of the disease (Figure 1).6, 7, 8, 9

Cutaneous neurofibromas.
Clinical description
The NIH Consensus Development Conference proposed the current clinical diagnostic criteria and nomenclature for neurofibromatosis 15 (Table 1).
Mosaic neurofibromatosis 1
More recently, mosaic forms of neurofibromatosis have been recognised. Somatic mutations arising early in embryogenesis produce generalised disease clinically indistinguishable from nonmosaic Nf1. Localised disease restricted to one part of the body results from later somatic mutations and occurs in one in 36 000 to one in 40 000 individuals.10
Diagnosis
The clinical diagnosis of Nf1 is evident by the age of 3 years in the majority of individuals.5, 6 The presence of UBOs on brain MRI has been advocated as a diagnostic criterion in young children with one feature of Nf1, but this age group needs general anaesthesia, thereby reducing the usefulness of the investigation. Diagnostic testing is available to confirm the diagnosis in individuals who fulfil the diagnostic criteria for Nf1, but are suspected of having the condition. Current mutation testing permits the identification pathogenic mutations in over 95% of Nf1 patients, using a combination of optimised protein truncation testing, fluorescent in situ hybridisation, direct sequencing, Southern blot analysis, and cytogenetic analysis.11 Prenatal testing is possible with foetal DNA extracted from chorionic villous sampling or from aminiocentesis. However, requests for prenatal testing are limited because of the inability to predict disease severity. Preimplantation diagnosis may be useful for a couple at risk for having a child with Nf1 and has been introduced in some centres.12
-
1)
Differential diagnosis.
-
2)
The major differential diagnoses of Nf1 are described in Table 2 13 Clinicians should be aware of individuals with segmental/mosaic Nf1 who present with six or more café au lait patches and skin-fold freckling in the affected area. It is important to distinguish between mosaic and generalised Nf1, as the former is a milder condition (see section on Genetic counselling).
-
3)
Homozygotes with mismatch repair syndromes can be misdiagnosed as having Nf1 as they have café au lait patches and an affected first-degree relative. However, in mismatch repair syndromes the affected relative is a sibling and the parents have a normal phenotype.
Description of disease manifestations
The Skin
Café au lait patches are present in 95% of Nf1 individuals and usually appear by the age of 3 years. Freckling develops in the majority of children in intertriginous areas and hypopigmented macules also occur.6 Xanthogranulomas are observed transiently in early childhood in 1–2% of patients as orange papules and have a putative link with juvenile chronic myeloid leukaemia.14
Neurofibromas and malignant peripheral nerve sheath tumours
Neurofibromas manifest as cutaneous (Figure 2), subcutaneous or plexiform lesions (Figure 3).6 Cutaneous neurofibromas cause itching and stinging and subcutaneous lesions often produce pain and neurological deficit from pressure on peripheral nerves.6 Neurofibromas rarely develop before age 7 years, usually emerge in late adolescence and frequently increase during pregnancy.6, 15 The number of tumours differs between individuals and the natural history is uncertain, with periods of rapid growth, followed by phases of quiescence. About 30% of Nf1 individuals have clinically visible plexiform neurofibromas,6 which are frequently congenital, often have a rich vascular supply and involve multiple nerve fascicles. Neurological deficit results from encroachment on surrounding structures and soft tissue and bony hypertrophy may coexist.16

Multiple cutaneous neurofibromas.

Benign plexiform neurofibroma of the right leg.
There is a 7–12% lifetime risk of developing an Nf1-associated malignant peripheral nerve sheath tumour (MPNST),17 which often arises within a pre-existing plexiform neurofibroma and metastases widely, often heralding a poor outcome. Individuals with subcutaneous neurofibromas are approximately three times more likely to have internal plexiform neurofibromas or MPNSTs than Nf1 sufferers with no subcutaneous lesions.18 These individuals warrant increased surveillance for MPNST as well as patients with plexiform neurofibromas in the brachial or lumbosacral plexus, a history of radiotherapy, a personal or family history of malignancy and Nf1 patients harbouring microdeletions of the Nf1 gene.9 Persistent pain, change in texture, rapid increase in size and neurological deficit associated with a neurofibroma are clinical features of malignancy (Figure 4).9 However, MPNSTs are difficult to diagnose, as similar symptoms are encountered in benign neurofibromas, magnetic resonance imaging (MRI) does not reliably distinguish MPNST and blind biopsy might miss the site of malignancy, because the tumours are heterogeneous.9

High-grade MPNST on the back.
Neurological complications
Cognitive impairment is the most common complication and patients present with low IQ, specific learning problems and behavioural difficulties.8 Abnormal executive function, impaired attention and language deficits have been observed. Most patients have an IQ in the low average range around 90 and mental retardation (IQ <70) is unusual.8 There is no evidence that cognitive ability improves with age.8
Unidentified bright objects (UBOs) have been identified as focal areas of high signal intensity on T2-weighted MRI.19 UBOs do not cause overt neurological deficit, they develop in the majority of children with Nf1, but most disappear in adulthood.8 It has been suggested that they represent delayed myelination or gliosis.20 A link between UBOs and cognitive dysfunction has been postulated, but the association has not been established universally.8 Neurological complications originate from malformations like aqueduct stenosis and tumours, including gliomas and ependymomas.6, 21 Gliomas occur ubiquitously in the central nervous system, chiefly in the optic pathways, brainstem and cerebellum.6, 21 Optic pathway gliomas (OPG) are often asymptomatic, but may cause visual impairment, squint, pupillary abnormalities, proptosis and hypothalamic dysfunction.22 They usually occur in the first 6 years of life and older individuals rarely develop symptomatic tumours.23
Skull and skeletal deformities may entail neurological sequelae – sphenoid wing dysplasia causes the temporal lobe to herniate into the orbit producing pulsating exophthalmos; severe, progressive scoliosis carries the risk of respiratory compromise and spinal cord compression.6
Neurofibromas produce neurological symptoms through pressure on peripheral nerves, spinal nerve roots and the spinal cord. Neurofibromatous neuropathy is characterised by a mild distal sensory–motor neuropathy associated with diffuse neurofibromatous change in thickened peripheral nerves.24
Neurofibromatous vasculopathy affects both arterial and venous circulations in the brain and manifestations include cerebral artery stenosis or occlusion, aneurysm and rupture.25 Cerebral haemorrhage accounts for 50% of deaths owing to cerebrovascular disease in Nf1.26 Multiple sclerosis and epilepsy (predominantly complex partial seizures) have been observed in association with Nf1, the latter probably arising from an underlying cortical dysgenesis.27, 28
Cardiovascular disease
Cardiovascular disease is a major cause of premature death in patients with Nf1.26.Essential hypertension is common and raised blood pressure owing to renal artery stenosis and phaeochromocytoma are observed. There is a higher than expected frequency of congenital heart disease in Nf1 individuals, particularly of valvular pulmonary stenosis.6, 25
Orthopaedic problems
Orthopaedic complications result from intrinsic defects of the skeletal system and from a disruption of bone structure maintenance.29 Bone mineral density is decreased in Nf1 patients, mainly in the load bearing parts. Pseudoarthrosis, a false joint in a long bone, affects 2% of Nf1 patients.6 Bowing of the affected long bone, most commonly the tibia, is apparent at birth or in the first few months of life, and fracture develops after trivial injury, with delayed healing. Scoliosis affects 10% of Nf1 patients and may be either idiopathic or dystrophic, the latter, progressive form developing after the age of 6 years, and rarely after the first decade.30
Genetics and molecular biology
Mutations in the Nf1 gene result in abnormal cell growth and proliferation and the formation of tumours. The Nf1 gene was identified on chromosome 17q11.2 and encodes a cytoplasmic protein neurofibromin, which is ubiquitously expressed with high levels of expression in the nervous system.2, 3, 4 Neurofibromin is related to the guanosine triphosphatase-activating proteins and has several known functions. Neurofibromin reduces cell proliferation by promoting the inactivation of p21RAS, which has a cardinal role in mitogenic intracellular signalling pathways.31 It also binds with microtubules and modulates adenylyl cyclase activity, which plays a cardinal role in cognition (see below).32 A common biochemical pathway for Nf1 and tuberous sclerosis has been identified recently. Neurofibromin regulates mTOR (mammalian target of rapamycin), a serine/threonine kinase that controls cell growth and division.33 It has been demonstrated that mTOR is activated in Nf1-deficient primary cells and Nf1-associated tumours. This activation of mTOR is dependent on ras and P13 kinase, which inactivate the TSC2 gene product tuberin, via AKT. Rapmaycin might have a therapeutic role in Nf1 as tumour cell lines derived from patients are sensitive to this mTOR inhibitor.33
Pathogenesis of neurofibromas
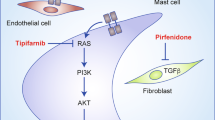
Neurofibromas comprise a mixture of Schwann cells, fibroblasts, perineurial cells, mast cells and axons embedded in an extracellular matrix.34 Schwann cells exhibit loss of Nf1 expression and the Schwann cell initiates neurofibroma growth. Zhu et al.35 ablated Nf1 function in mouse Schwann cells using a conditional (cre/lox) allele and reported a novel observation in a tumour suppressor syndrome. The mice developed neurofibromas identical to those seen in humans, only when null Schwann cells were present on an Nf1 heterozygote background and not in an otherwise normal mouse. They observed increased numbers of mast cells and hypothesised that they might contribute to neurofibroma formation.35 RAS-GTP levels are increased in some neurofbroma Schwann cells but not in fibroblasts, suggesting that other genetic and epigenetic events are required to produce neurofibromas.36 Candidates include expression of epidermal and vascular endothelial growth factors and their receptors and matrix metalloproteinases.37, 38 Normal cellular relationships within the perineurium are contingent upon closely regulated signalling between Schwann cells, axons, fibroblasts and perineurial cells. Impaired signalling between the components of neurofibromas might promote neurofibroma development.
MPNST
MPNST from Nf1 individuals and sporadic MPNST both exhibit loss of Nf1 expression.39 However, malignant change demands additional genetic events that inactivate key cell cycle regulators, including p53, p16 and p27-kip1.40, 41 Furthermore, mice with targeted mutations of the Nf1 and p53 genes develop MPNST, when these genes are inactivated.42
OPG and astrocytomas
The molecular pathogenesis of OPG has been difficult to unravel because these indolent tumours rarely require surgery. Inactivation of Nf1 is found in Nf1-associated pilocytic astrocytomas but not in sporadic tumours, in contrast to MPNST. The natural history of OPG formation has been studied in a mouse model with immunohistochemistry and diffusion tensor imaging.43 A 2-month-old mice developed OPG that were demonstrated as contrast enhancing tumours on MRI.43 Microglial cell infiltration and new vessel formation were observed in the period before tumour formation and the OPG exhibited expression of proteins associated with astroglial precursors.43 Recent investigations have shown that loss of neurofibromin in astrocytes leads to the activation of the KRAS isoform in astrocytes and the mTOR–S6 kinase pathway, leading to increased cell proliferation and protein translation.44 Rapacmycin can block astrocyte proliferation in vitro, thereby forming a potential therapeutic target for Nf1-related brain tumours.44
Cognition
The adenylyl cyclase pathway has been implicated in deficits in learning and memory in the Drosophila melanogaster as the deficits were corrected by protein kinase A activation.32Studies in mice that are heterozygous for the Nf1 mutation, suggest that cognitive impairment in Nf1 is linked with excessive RAS activity and increased GABA inhibition in the hippocampus.45 Furthermore, the learning problems and the GABA inhibition are reversed by a reduction in RAS, implying that farnesyl transferase inhibitor drugs might play a therapeutic role in cognitive impairment in Nf1.45 The cholesterol lowering drug Lovastatin inhibits p21RAS/mitogen activated protein kinase and a recent study demonstrated that the drug reverses learning and attention deficits in a mouse model of Nf1.46 Currently, Lovostatin is being investigated as a potential treatment for cognitive impairment in children with Nf1.
Management
Children and adults with Nf1 should be examined yearly by a clinician conversant with Nf1 and its complications. Annual assessment of the skin and blood pressure should be performed in both groups. Children require monitoring of the spine, growth, cognitive development and school progress. Our practice is to perform a visual examination with visual fields, acuity, colour vision developmental maturity allows, and then annual visual assessment is undertaken by the optician.
Neurofibromas
The mainstay of treatment for cutaneous neurofibromas is surgical removal, notwithstanding occasional hypertrophic scarring, and carbon dioxide laser may be indicated for small, superficial lesions. About 75% of neurofibromas express progesterone receptors, suggesting a potential therapeutic role form antiprogesterone therapy.47 Plexiform neurofibromas are often difficult to remove because of impingement on soft tissue and on major nerves, the tendency of some lesions to bleed profusely and the possibility of re-growth. Radiotherapy is contraindicated because of the risk of malignant transformation. Several chemotherapy trials have been conducted to treat plexiform neurofibromas, including antihistamines, maturation agents, and antiangiogenesis drugs.48 A randomised placebo-controlled trial is in progress to assess an oral farnesyltransferase inhibitor and the antifibrotic agent pirfenidone is being evaluated in adults with progressive plexiform and spinal neurofibromas.48
MPNST
Clinicians and patients need to be alert to the symptoms of malignant change in a neurofibroma and prompt referral to a specialist NF/sarcoma unit should be ensured.9 The use of 18fluorodeoxyglucose positron emission tomography facilitates early diagnosis of MPNST.9 The optimum treatment is surgical excision of the tumour with tumour-free margins.9 Radiotherapy improves local control for incompletely excised tumours and for intermediate and high-grade MPNSTs. Chemotherapy with ifosfamide and doxorubicin is palliative in metastatic disease and might play a role in reducing the size of a tumour before surgery.9
Orthopaedic problems
Most patients with pseudoarthrosis require surgery and amputation is necessary in severe cases. Recent research suggests that psuedoarthrosis might be treated using autograft mesenchymal stem cells from the healthy iliac crest.49 The dystrophic form of scoliosis is characterised by a rapidly progressive curve, requiring early spinal fusion.6
OPG
Chemotherapy with vincristine and cisplatinum is the optimum treatment for progressive symptomatic OPG, but radiotherapy is not recommended for young children because of neuropsychiatric, endocrinological, and vascular complications.50 Screening for asymtpomatic OPG is not advocated, as it does not influence the need for treatment or outcome.
Cognitive impairment
Clinicians, teachers and parents should recognise the possibility of educational and behavioural problems in children with Nf1, so that structured remedial teaching can be offered at an early stage.8 Children with attention deficit may respond well to dexamphetamine or methylphenidate under skilled supervision.8
Genetic counselling
The clinical diagnosis is clearcut in most individuals with Nf1 and where there is doubt the patients should be seen in a specialist clinic before mutation testing is undertaken. Approximately 50% of patients have Nf1 as a new mutation, while an affected parent has a 50% chance of having a child with the disease.6
The risk of an individual with mosaic Nf1 passing on generalised disease to an offspring is small but unquantifiable, as it depends on the percentage of body that is affected.10 Apparently normal parents of an affected child should be examined carefully for the presence of mosaic Nf1. If the parents are normal, the risk of recurrence is probably only barely above the background risk of 1/6000. There is little evidence for the high levels of gonadal mosaicism seen in other conditions such as tuberous sclerosis. As the phenotype of Nf1 is variable, it is difficult to predict the risks of complications in any one individual. A mildly affected patient might have a child with severe disease, and the converse may be true for a parent with severe disease. The chance of an individual with Nf1 having a severely affected child is 8%. Individuals with Nf1 and whole gene deletions have been described and they have distinct phenotype with dysmorphism, significant cognitive impairment and large numbers of cutaneous neurofibromas.51
There is marked clinical heterogeneity between individuals with Nf1, even within families. Recent research suggests that individuals with Nf1 microdeletions might be at higher risk of developing MPNST.52 This finding has not been universally established and a larger cohort needs to be assessed to verify the premise (Upadhyaya M et al, personal communication). It has been hypothesised that the variation in clinical expression in Nf1 is due to the nature, timing or location of ‘second hit mutations’ at the Nf1 locus, to somatic mosaicism or to the presence of modifying genes.53 The presence of modifying genes is supported by frequent observations that identical Nf1 mutations give rise to different clinical phenotypes.
Conclusions
Nf1 is a complex neurocutaneous disease requiring supervision and management by an expert multidisciplinary team. Elucidation of the pathogenesis of Nf1 and advances in treatment will be enhanced by close collaboration between clinicians and scientists and the ready availability of meticulously documented clinical data, blood and tumour tissue for research. The current advances in molecular biology provide the hope of targeted therapy for this distressing disease. However, reliable clinical and radiographic outcome measures must be developed to assess the potential benefits of any future drug trials in patients.
References
Further Reading
Friedman JM, Gutmann DH, MacCollin M, Riccardi VM (eds): Neurofibromatosis: Phenotype, Natural History and Pathogenesis. Baltimore, MD: Johns Hopkins University Press, 1999.
Ward BA, Gutmann DH : Nf1: from Lab bench to Bedside. Paediatr Neurol 2005; 32: 221–228.
Ducatman B, Scheithauer B, Piepgras D et al: Malignant peripheral nerve sheath tumours: a clinicopathological study of 120 cases. Cancer (Philadelphia) 1986; 57: 2006–2021.
Ferner R, Lucas JD, O’Doherty et al: Evaluation of 18-fluorodeoxyglucose positron emission tomography (18FDGPET) in the detection of malignant peripheral nerve sheath tumours arising from within plexiform neurofibromas in neurofibromatosis 1. J Neurol Neurosurg Psychiat 2000; 68: 353–357.
Xu HM, Gutmann DH : Merlin differentially associates with microtubule and actin cytoskeleton. J Neurosci Res 1998; 51: 403–415.
Costa RM, Silva AJ : Mouse models of neurofibromatosis type 1: bridging the GAP. Trends Mol Med 2003; 9: 19–23.
Mautner VF, Kluwe L, Thakker SD et al: Treatment of ADHD in neurofibromatosis type 1. Dev Med Child Neurol 2002; 44: 164–170.
Hyman SL, Shores A, North KN : The nature and frequency of cognitive deficits in children with neurofibromatosis 1. Neurol 2005; 11: 1037–1044.
Evans DGR, Huson SM, Donnai D et al: A clinical study of type 2 neurofibromatosis. Q J Med 1992; 84: 603–618.
Evans DGR, Baser ME, O’Reilly B et al: Management of the patient and family with neurofibromatosis. 2: A consensus conference statement. Br J Neurosurg 2005; 19: 5–12.
Ramesh V : Merlin and ERM proteins in Schwann cells, neurons and growth cones. Nat Rev Neurosci 2004; 5: 462–470.
MacCollin M, Chiocca EA, Evans DG et al: Diagnostic criteria for schwannomatosis. Neurology 2005; 64: 1838–1845, (Resources, The Children's Tumour Foundation, 95, Pine Street, 16th Floor, New York, NY 10005, USA, E-mail: info@ctf.org, www.ctf.org).
References
Huson SM, Compston DAS, Clark P et al: A genetic study of von Recklinghausen neurofibromatosis in south east Wales. 1. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet 1989; 26: 704–711.
Viskochil D, Buchberg AN, Xu G et al: Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 1990; 62: 1887–1892.
Wallace MR, Marchuk DA, Anderson LB et al: Type 1 neurofibromatosis gene: identification of a larger transcript disrupted in three NG1 patients. Science 1990; 249: 181–186.
Xu GF, O’Connell P, Viskochil D et al: The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 1990; 62: 599–608.
National Institutes of Health Consensus Development Conference Statement: Neurofibromatosis. Arch Neurol (Chicago) 1988; 45: 575–578.
Huson SM, Harper PS, Compston DAS : Von Recklinghausen neurofibromatosis: clinical and population study in South East Wales. Brain 1988; 111: 55–81.
Friedman JM, Arbiser J, Epstein JA et al: Cardiovascular disease in neurofibromatosis 1: report of the Nf1 cardiovascular task force. Gen Med 2002; 4: 105–111.
North KN, Riccardi V, Samango-Sprouse C et al: Cognitive function and academic performance in neurofibromatosis. 1: Consensus statement from the Nf1 Cognitive Disorders Task Force. Neurology 1997; 48: 1121–1127.
Ferner RE, Gutmann DH : International consensus statement on malignant peripheral nerve sheath tumours in neurofibromatosis 1. Cancer Res 2002; 62: 1573–1577.
Ruggieri M, Huson SM : The clinical and diagnostic implications of mosaicism in the neurofibromatoses. J Neurol 2001; 56: 1433–1443.
Messiaen LM, Callens T, Mortier G et al: Exhaustive mutation analysis of the Nf1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat 2000; 15: 541–555.
Verlinsky Y, Rechitsky S, Verlinsky O et al: Preimplantation diagnosis for neurofibromatosis. Reprod Biomed Online 2002; 4: 218–222.
Viskochil D, Carey JC : Alternate and related forms of the neurofibromatoses; in Huson SM, Hughes RACH (eds): The Neurofibromatoses. London: Chapman & Hall, pp 445–474.
Zvulunov A, Bark Y, Mejer A : Juvenile xanthogranulomas, neurofibromatosis and juvenile chronic myelogenous leukaemia. World statistical analysis. Arch Dermatol 1995; 131: 904–908.
Dugoff L, Sujansky E : Neurofibromatosis type 1 and pregnancy. Am J Med Genet 1996; 66: 7–10.
Korf B, Huson SM, Needle M et al: Report of the working group on neurofibroma. Nat Neurofibromatosis Found Inc 1997; 4–27.
Evans DG, Baser ME, McGaughran J et al: Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 2002; 39: 311–314.
Tucker T, Wolkenstein P, Revuz J et al: Association between benign and malignant peripheral nerve sheath tumours in Nf1. Neurol 2005; 65: 205–211.
Bognanno JR, Edwards MK, Lee TA et al: Cranial MR imaging in neurofibromatosis. Am J Radiol 1988; 151: 381–388.
DiPaolo DP, Zimmerman RA, Rorke LB et al: Neurofibromatosis type 1: pathologic substrate of high-signal-intensity foci in the brain. Radiology 1995; 195: 721–724.
Créange A, Zeller J, Rostaing-Rigattieri S et al: Neurological complications of neurofibromatosis type 1 in adulthood. Brain 1999; 122: 473–481.
Listernick R, Louis DN, Packer RJ et al: Optic pathway gliomas in children with neurofibromatosis type. 1: Consensus statement from the Nf1 optic pathway glioma study. J Paediatr 1994; 125: 63–66.
Listernick R, Ferner RE, Piersall L et al: Late-onset optic pathway tumours in children with neurofibromatosis 1. Neurology 2004; 63: 1944–1946.
Ferner RE, Hall SM, Hughes RAC et al: Neurofibromatous neuropathy in neurofibromatosis 1. J Med Genet 2004; 41: 837–841.
Friedman JM, Arbiser J, Epstein JA et al: Cardiovascular disease in neurofibromatosis. 1: A report of the Nf1 Cardiovascular Task Force. Genet Med 2003; 4: 105–111.
Rasmussen SA, Yang Q, Friedman JM : Mortality in neurofibromatosis. 1: An analysis using US death certificates. Am J Hum Genet 2001; 68: 1110–1118.
Ferner RE, Hughes RAC, Johnson MR : Neurofibromatosis 1 and multiple sclerosis. J Neurol Neurosurg Psychiat 1995; 56: 492–495.
Vivarelli R, Grosso S, Calabrese F et al: Epilepsy in neurofibromatosis 1. J Child Neurol 2003; 18: 338–342.
Kuorilehto T, Poyhonen M, Bloigu R et al: Decreased bone mineral density and content in neurofibromatosis type 1: Lowest local values are located in the load-carrying parts of the body. Osteoporosis Int 2005; 16: 928–936.
Crawford Jr AH, Bagamery N : Osseous manifestations of neurofibromatosis in childhood. J Pediatr Orthop 1986; 6: 72–88.
Gutmann DH, Wood DL, Collins FS : Identification of the neurofibromatosis 1 gene product. Proc Natl Acad Sci USA 1991; 88: 9658–9762.
Guo HF, Tong J, Hannan F et al: A neurofibromatosis -1- regulated pathway is required for learning in Drosophila. Nature 2000; 403: 895–898.
Johannessen CM, Reczek EE, James MF et al: The Nf1 tumour suppressor critically regulates TSC2 and m TOR. Proc Natl Acad Sci USA 2005; 102: 8573–8578.
Kimura M, Kamata Y, Matsumoto K et al: Electron microscopical study on the tumour of von Recklinghausen's neurofibromatosis. Pathol Jpn 1974; 24: 79–91.
Zhu Y, Ghosh P, Charnay P et al: Neurofibromas in Nf1: Schwann cell origin and role of tumour environment. Science 2002; 296: 920–922.
Sherman LS, Atit R, Rosenbaum T, Cox AD, Ratner N : Single cell RAS-GTP analysis reveals altered RAS activity in a subpopulation of neurofibroma Schwann cells but not fibroblasts. J Biol Chem 2000; 275: 30740–30745.
DeClue JE, Heffelfinger S, Benvenuto G et al: Epidermal growth factor receptor expression in neurofibromatosis type 1-related tumours and Nf1 animal models. J Clin Invest 2000; 105: 1233–1241.
Muir D : Differences in proliferation and invasion by normal, transformed and Nf1 Schwann cell cultures are influenced by matrix metalloproteinase expression. Clin Exp Metast 1995; 13: 303–314.
Guha A, Lau N, Huvar I et al: RAS-GTP levels are elevated in human peripheral nerve tumours. Oncogene 1996; 12: 507–513.
Nielsen GP, Stemmer-Rachamimov AO, Ino Y et al: Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKNA/p16 inactivation. Am J Pathol 1999; 155: 1879–1884.
Kourea HP, Cordon-Cardo C, Dudas M et al: The emerging role of p27kip in malignant transformation of neurofibromas. Am J Pathol 1999; 155: 1885–1891.
Cichowski K, Shih TS, Schmitt E et al: Mouse models of tumour development in neurofibromatosis type 1. Science (Washington, DC) 1999; 286: 2172–2176.
Bajenaru ML, Garbow JR, Perry A et al: Natural history of neurofibromatosis 1-associated optic nerve glioma in mice. Ann Neurol 2005; 57: 119–127.
Dasgupta B, Yi Y, Chen DY et al: Proteomic analysis reveals hyper-activation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumours. Cancer Res 2005; 1: 2755–2760.
Costa RM, Federov NB, Kogan JH et al: Mechanisms for the learning deficits in a mouse model of neurofibromatosis 1. Nature 2002; 415: 526–530.
Li W, Kushner SA, Brown RA et al: The HMG-CoA reductase inhibitor lovostatin reverses the learning and attention deficits in a mouse model of neurofibromatosis 1. Curr Biol 2005; 15: 1961–1967.
McLaughlin ME, Jacks T : Progesterone receptor expression in neurofibromas. Cancer Res 2003; 63: 752–755.
Packer RJ, Gutmann DH, Rubenstein A et al: Plexiform neurofibromas in N1: toward biologic-based therapy. Neurology 2002; 58: 1461–1470.
Kitoh H, Kitakoji T, Tsuchiya H et al: Transplantaton of marrow-derived mesenchymal stem cells and platelet-rich plasma during distraction osteogenesis – a preliminary result of three cases. Bone 2004; 35: 892–898.
Packer RJ, Alter J, Allen J et al: Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg 1997; 86: 747–754.
Tonsgard JH, Yelavarthi KK, Cushner S et al: Do Nf1 gene deletions result in a characteristic phenotype? Am J Med Genet 1997; 73: 80–86.
De Raedt T, Brems H, Wolkenstein P et al: Elevated risk for MPNST in Nf1 microdeletion patients. Am J Hum Genet 2003; 72: 1288–1292.
Easton DF, Ponder MA, Huson SM et al: An analysis of variation in expression of neurofibromatosis type 1 (Nf1): evidence for modifying genes. Am J Hum Genet 1993; 53: 305–313.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ferner, R. Neurofibromatosis 1. Eur J Hum Genet 15, 131–138 (2007). https://doi.org/10.1038/sj.ejhg.5201676
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201676
Keywords
This article is cited by
-
Perinatal folate levels do not influence tumor latency or multiplicity in a model of NF1 associated plexiform-like neurofibromas
BMC Research Notes (2023)
-
Sex- and age-related differences in autistic behaviours in children with neurofibromatosis type 1
Journal of Autism and Developmental Disorders (2023)
-
Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966
Orphanet Journal of Rare Diseases (2020)
-
Knockdown of HMGA2 regulates the level of autophagy via interactions between MSI2 and Beclin1 to inhibit NF1-associated malignant peripheral nerve sheath tumour growth
Journal of Experimental & Clinical Cancer Research (2019)
-
Self-organizing neuruloids model developmental aspects of Huntington’s disease in the ectodermal compartment
Nature Biotechnology (2019)
